 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
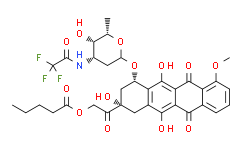




















| Cas No.: | 56124-62-0 |
| Chemical Name: | Valrubicin |
| Synonyms: | Valrubicin;[2-Oxo-2-[(2S,4S)-2,5,12-trihydroxy-4-[5-hydroxy-6-methyl-4-[(2,2,2-trifluoroacetyl)amino]oxan-2-yl]oxy-7-methoxy-6,11-dioxo-3,4-dihydro-1H-tetracen-2-yl]ethyl] pentanoate;AD-32;ad32;antibioticad32;N-(trifluoroacetyl)adriamycin 14-valerate (AD 32);N-trifluoroacetyladriamycin-14-valerate;N-trifluoroacetyldoxorubicin-14-valerate;trifluoroacetyl-adriamyci14-valerate;Valstar;Vinorelbine (BICINS );Pentanoicacid,2-[(2S,4S)-1,2,3,4,6,11-hexahydro-2,5,12-trihydroxy-7-methoxy-6,11-dioxo-4-[[2,3,6-trideoxy-3-[(trifluoroacetyl)amino]-a-L-lyxo-hexopyranosyl]oxy]-2-naphthacenyl]-2-oxoethylester (9CI);Pentanoic acid,2-[1,2,3,4,6,11-hexahydro-2,5,12-trihydroxy-7-methoxy-6,11-dioxo-4-[[2,3,6-trideoxy-3-[(trifluoroacetyl)amino]-a-L-lyxo-hexopyranosyl]oxy]-2-naphthacenyl]-2-oxoethylester, (2S-cis)-;AD 32;Antibiotic AD 32;N-Trifluoroacetyladriamycin14-valerate;N-Trifluoroacetyldoxorubicin 14-valerate;NSC 246131;Trifluoroacetyladriamycin 14-valerate |
| SMILES: | O([C@]1([H])O[C@@H](C)[C@@H](O)[C@@H](NC(=O)C(F)(F)F)C1)[C@H]1C[C@](O)(C(=O)COC(=O)CCCC)CC2C(=C3C(C4=CC=CC(OC)=C4C(=O)C3=C(O)C1=2)=O)O |
| Formula: | C34H36F3NO13 |
| M.Wt: | 723.643761634827 |
| Purity: | >98% |
| Sotrage: | 2 years -20°C Powder, 2 weeks 4°C in DMSO, 6 months -80°C in DMSO |
| Description: | Valrubicin is a chemotherapy agent, inhibits TPA- and PDBu-induced PKC activation with IC50s of 0.85 and 1.25 μM, respectively, and has antitumor and antiinflammatory activity. |
| Target: | TPA-activated PKC:0.85 μM (IC50) PDBu-activated PKC:1.25 μM (IC50) |
| In Vivo: | Valrubicin (3, 6, or 9 mg) reduces tumor growth at week 3 by intratumoral jection in hamster. Valrubicin (6 mg) combined with minimally cytotoxic irradiation (150, 250, or 350 cGy) causes significant tumor shrinkage in hamster[2]. Valrubicin (0.1 μg/μL) significantly reduces the number of infiltrating neutrophils in biopsies challenged with TPA at 24 h and attenuates chronic inflammation in mice. Valrubicin also decreases the expression levels of inflammatory cytokines in the acute model[3]. |
| In Vitro: | Valrubicin (AD 32) is a chemotherapy agent, inhibits TPA- and PDBu-induced PKC activation with IC50s of 0.85 and 1.25 μM, respectively. Valrubicin inhibits the binding of [3H]PDBu to PKC. Therefore, Valrubicin competes with the tumor promoter for the PKC binding site and prevents the latter from both interacting with the phospholipid and binding to PKC[1]. Valrubicin shows cytotoxic activity against squamous cell carcinoma (SCC) cell line colony formation, with IC50s and IC90s of 8.24 ± 1.60 μM and 14.81 ± 2.82 μM for UMSCC5 cells, 15.90 ± 0.90 μM, 29.84 ± 0.84 μM for UMSCC5/CDDP‡ cells, and 10.50 ± 2.39 μM, 19.00 ± 3.91 μM for UMSCC10b cells, respectively. Moreover, Valrubicin in combination with radiation enhances the cytotoxicity[2]. |
| Cell Assay: | UMSCC5 cells exposed to Valrubicin (2 μM for 3 h), a single dose of radiation (400 cGy), or the combined treatment are cultured for a further 12, 24, or 48 hours. At these times, the cells are collected by trypsinization (0.25%), washed in phosphate-buffered saline (PBS), and fixed at 5 × 106 cells/mL with 95% ethanol. Cells are incubated with ribonuclease (50 μg; 70-90 Kunitz units/mg for 30 min), and the resulting pellet resuspended in and incubated with propidium iodide (0.05 mg/mL for 10 min). The DNA content of the samples is determined by flow cytometry according to standard technique[2]. |
| Animal Administration: | Hamsters[2] Hamsters with cheek pouch tumors of 100 mm2 are randomly assigned to one of five treatment groups. Momentarily anesthetized animals each receives once a week × 3 injections (27 g × 0.5-inch needle: 0.1 mL administered slowly to the base of the lesion) of Valrubicin (3, 6, or 9 mg) or drug vehicle (Cremophor: alcohol;1:1 by volume; NCl diluent 12). A further group of animals receives anesthesia but no direct tumor treatment (control). Individual tumor sizes are measured with calipers at weekly intervals for 4 weeks, at which time the animals are sacrificed[2]. |
| References: | [1]. Chuang LF, et al. Activation of human leukemia protein kinase C by tumor promoters and its inhibition by N-trifluoroacetyladriamycin-14-valerate (AD 32). Biochem Pharmacol. 1992 Feb 18;43(4):865-72. [2]. Wani MK, et al. Rationale for intralesional valrubicin in chemoradiation of squamous cell carcinoma of the head and neck. Laryngoscope. 2000 Dec;110(12):2026-32. [3]. Hauge E, et al. Topical valrubicin application reduces skin inflammation in murine models. Br J Dermatol. 2012 Aug;167(2):288-95. |