 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
| Cat. No. | Product Name | Field of Application | Chemical Structure |
|---|---|---|---|
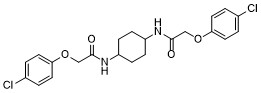
| DC70309 | cis-ISRIB Featured |
cis-ISRIB is a weak inhibitor of PERK signaling compared to trans-ISRIB , potently reverses the effects of eIF2α phosphorylation with IC50 of 600 nM.ISRIB reduces the viability of cells subjected to PERK-activation by chronic endoplasmic reticulum stress, displays significant enhancement in spatial and fear-associated learning.ISRIB is a potent inhibitor of the integrated stress response (ISR)integrated stress response (ISR).
More description
|
|
| DC8453 | Talazoparib(BMN-673)tosylate Featured |
Talazoparib, also known as BMN-673 and MDV-3800, is an orally bioavailable inhibitor of the nuclear enzyme poly(ADP-ribose) polymerase (PARP) with potential antineoplastic activity (PARP1 IC50 = 0.57 nmol/L). Talazoparib acts as an inhibitor of poly ADP ribose polymerase(PARP) which aids in single strand DNA repair. Cells that have BRCA1/2 mutations are susceptible to the cytotoxic effects of PARP inhibitors because of an accumulation of DNA damage. Talazoparib is theorized to have a higher potency than olaparib due to the additional mechanism of action called PARP trapping. PARP trapping is the mechanism of action where the PARP molecule is trapped on the DNA, which interferes with the cells ability to replicate. Talazoparib is found to be ~100 fold more efficient in PARP trapping than olaparib.
More description
|

|
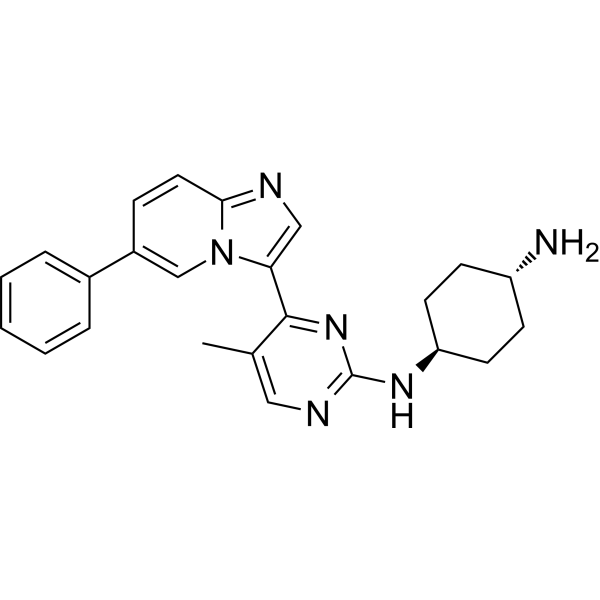
| DC70298 | CDDD11-8 Featured |
CDDD11-8 is a potent CDK9 inhibitor co-targeting FLT3-ITD with Ki values of 8 and 13 nM, respectively.CDDD11-8 displays excellent kinome selectivity in a panel of 369 human kinases.CDDD11-8 displays antiproliferative activity against leukemia cell lines, and particularly potent effects against MV4-11 and MOLM-13 cells, which are known to harbor the FLT3-ITD mutation and mixed lineage leukemia (MLL) fusion proteins.CDDD11-8 causes a robust tumor growth inhibition by oral administration in animal xenografts, induces tumor regression at dose of 125 mg/kg.
More description
|
|
| DC11199 | CZh226 Featured |
CZh226 hydrochloride (CZh-226) is a potent, selective inhibitor of p21-activated kinase 4 (PAK4) with Ki of 9 nM, displays 346-fold selectivity over PAK-1.
More description
|

|
| DC7076 | AZD-7762 Featured |
AZD7762 is a potent and selective inhibitor of Chk1 with IC50 of 5 nM; equally potent against Chk2 and less potent against CAM, Yes, Fyn, Lyn, Hck and Lck.
More description
|

|
| DC11470 | Belotecan hydrochloride(CKD-602) Featured |
Belotecan hydrochloride (CKD-602 hydrochloride), a Topoisomerase I inhibitor, is a synthetic and water-soluble camptothecin derivative.
More description
|

|
| DC8111 | Idarubicin Hydrochloride Featured |
Idarubicin Hcl is an anthracycline antibiotic and a DNA topoisomerase II (topo II) inhibitor.
More description
|
.gif)
|
| DC2014 | Flavopiridol Featured |
Flavopiridol competes with ATP to inhibit CDKs including CDK1, CDK2, CDK4 and CDK6 with IC50 of ~ 40 nM, and CDK7 with IC50 of 300 nM.
More description
|

|
| DC46399 | ZN-c3 Featured |
ZN-c3 is an orally active, highly potent and selective Wee1 inhibitor (IC50=3.9 nM). ZN-c3 can be used for the research of cancer.
More description
|
.png)
|
| DC10611 | Trabectedin Featured |
Trabectedin (Ecteinascidin-743 or ET-743) is a novel antitumour agent of marine origin with potent antitumour activity both in vitro and in vivo.
More description
|

|
| DC44851 | CHAPS Featured |
CHAPS is a zwitterionic surfactant that decreases the sequence specificity of the nucleosome.,nucleosome [1]()
More description
|

|
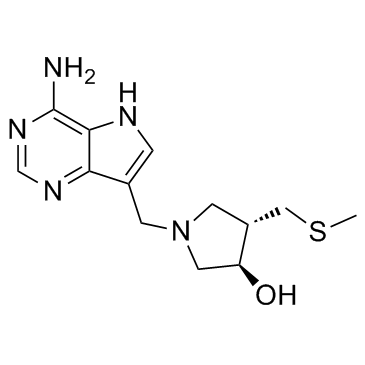
| DC10332 | Methylthio-DADMe-Immucillin A; MTDIA Featured |
MT-DADMe-ImmA is an inhibitor of human 5'-methylthioadenosine phosphorylase (MTAP) with a Ki of 90 pM.
More description
|
|
| DC8861 | 2'-Deoxythioguanosine Featured |
6-thio-dG is a nucleoside analog and telomerase substrate.
More description
|

|
| DCAPI1525 | 5-Azacytidine Featured |
5-Azacytidine is a potent growth inhibitor and a cytotoxic agent. 5-Azacytidine acts as a demethylating agent by inhibiting DNA methyltransferase (Dnmt), forming covalent adducts with cellular DNMT1 depleting enzyme activity. 5-Azacytidine also improves t
More description
|

|
| DC7114 | Dinaciclib (SCH727965) Featured |
Dinaciclib (SCH727965) is a novel and potent CDK inhibitor for CDK2, CDK5, CDK1 and CDK9 with IC50 of 1 nM, 1 nM, 3 nM and 4 nM, respectively.
More description
|

|
| DC9774 | LEE011 succinate Featured |
LEE011 succinate is an orally available cyclin-dependent kinase (CDK) inhibitor targeting cyclin D1/CDK4 and cyclin D3/CDK6 cell cycle pathway, with potential antineoplastic activity.
More description
|
.gif)
|
| DC10318 | Acelarin Featured |
Acelarin (NUC-1031) is a ProTide transformation and enhancement of the widely-used nucleoside analogue, gemcitabine.
More description
|

|
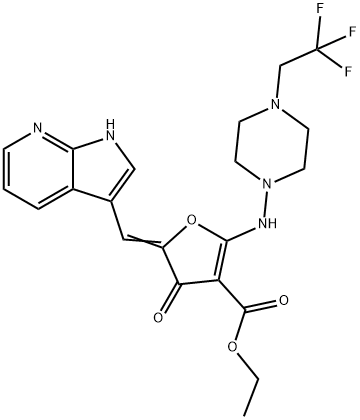
| DC28660 | Cdc7-IN-6 Featured |
Cdc7-IN-6 (compound I-D) is a potent Cdc7 kinase inhibitor (IC50=4 nM), extracted from patent WO2019165473A1, compound I- D, has anti-tumor activity.
More description
|
|
| DC9576 | Niraparib tosylate Featured |
MK-4827(Niraparib) tosylate is a selective inhibitor of PARP1/PARP2 with IC50 of 3.8 nM/2.1 nM; with great activity in cancer cells with mutant BRCA-1 and BRCA-2; >330-fold selective against PARP3, V-PARP and Tank1.
More description
|

|
| DC40472 | γ-Amanitin Featured |
γ-Amanitin an ADC cytotoxin and isolated from the?mushroom. γ-Amanitin inhibits?RNA polymerase II and disrupts synthesis of?mRNA. γ-Amanitin shows similar effects to α-Amanitin and β-Amanitin .
More description
|

|
| DC7448 | KU-60019 Featured |
KU-60019 is an improved analogue of KU-55933, with IC50 of 6.3 nM for ATM.
More description
|

|
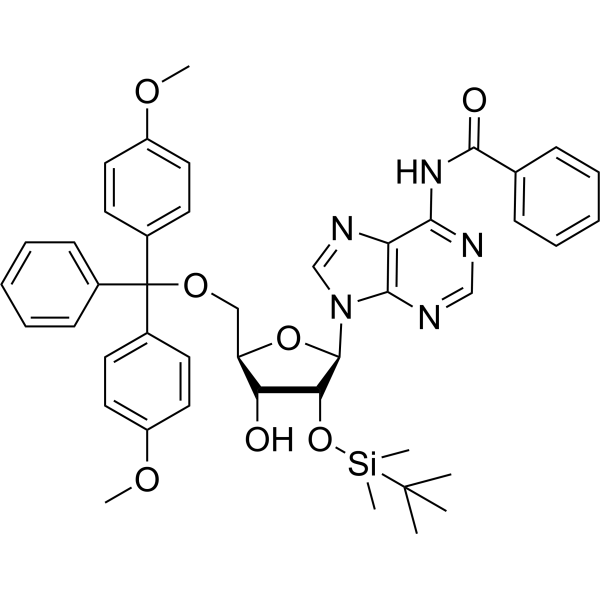
| DC44975 | 5'-O-DMT-2'-O-TBDMS-N-Bz-Adenosine Featured |
5'-O-DMT-2'-O-TBDMS-N-Bz-Adenosine is an adenosine derivative and can be used as an intermediate for oligonucleotides synthesis.
More description
|
|
| DC11163 | AST-3424 Featured |
AST-3424 (OBI-3424, TH-3424) is a first-in-class, novel prodrug that selectively targets cancers overexpressing the enzyme AKR1C3, OBI-3424 is a highly selective prodrug that is converted by AKR1C3 to a DNA alkylating agent.
More description
|

|
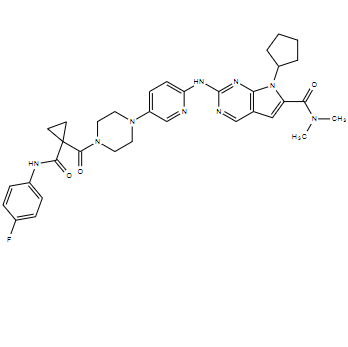
| DC55130 | CDK4 inhibitor compound 12 Featured |
CDK4 inhibitor compound 12 is a novel inhibitor of CDK4 with the activity 97 μM.
More description
|
|
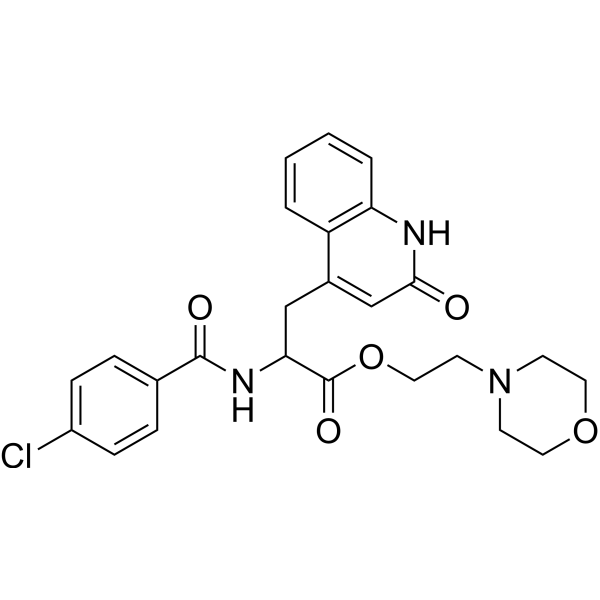
| DC47048 | Rebamipide mofetil Featured |
Rebamipide mofetil is an orally active prodrug of Rebamipide (OPC12759). Rebamipide is a mucoprotective agent. Rebamipide induces COX-2 expression, increases PGE2 levels, and enhances gastric mucosal defense in a COX-2-dependent manner.
More description
|
|
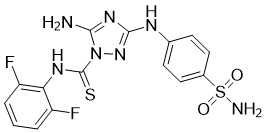
| DC28292 | Cdk1/2 Inhibitor III Featured |
Cdk1/2 Inhibitor III is a selective Cdk1/2 inhibitor, with an IC50 of 2.1 μM for CDK1/cyclin B.
More description
|
|
| DC46283 | Sibiromycin Featured |
Sibiromycin is a naturally produced glycosylated pyrrolobenzodiazepines (PBDs). Sibiromycin is also a potent antitumor antibiotic that binds covalently to DNA in the minor groove at the NH2 of guanine.
More description
|

|
| DC40300 | Illudin M Featured |
Illudin M is a cytotoxic fungal sesquiterpene that can be isolated from the culture medium of Omphalotus olearius mushrooms. Illudin M can alkylate DNA. Illudin M has anti-tumor activities.
More description
|

|
| DC39082 | BCH001 Featured |
BCH001 is a specific PAPD5 inhibitor that restores telomerase activity and telomere length in dyskeratosis congenita (DC) patient induced pluripotent stem cells. PAPD5 is a noncanonical poly(A) polymerase with an unusual RNA-binding motif.
More description
|

|
| DC45551 | LY3405105 Featured |
LY3405105 is an orally active CDK7 inhibitor with an IC50 of 92.8 nM. LY3405105 shows potential antineoplastic activity.
More description
|

|