 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
| Cat. No. | Product Name | Field of Application | Chemical Structure |
|---|---|---|---|
| DC77943 | Nivudirsen |
Nivudirsen is an antisense oligonucleotide that can promote the synthesis of functional dystrophin protein.
More description
|

|
| DC77942 | Nivudirsen sodium |
Nivudirsen sodium is an antisense oligonucleotide that can promote the synthesis of functional dystrophin protein.
More description
|
|
| DC77937 | FBnG-amino-PEG3-C2-azido |
FBnG-amino-PEG3-C2-azido is a tag-linker conjugate that incorporates a degradation tag FBnG and a glycol linker (Amino-PEG3-C2-Azido). FBnG-amino-PEG3-C2-azido can be used for synthesis of GPX4-AUTAC
More description
|
|
| DC77936 | Duazomycin sodium |
Duazomycin (Duazomycin A) sodium is a glutamine antagonist. Duazomycin sodium can significantly enhance the effectiveness of 6-Mercaptopurine (6-MP) in experimental allergic encephalomyelitis (EAE) without increasing toxicity.
More description
|
|
| DC77935 | Movronersen |
Movronersen is an antisense oligonucleotide targeted to α-synuclein.
More description
|
|
| DC77934 | Movronersen sodium |
Movronersen sodium is an antisense oligonucleotide targeted to α-synuclein.
More description
|
|
| DC77933 | Mivelsiran |
Mivelsiran is a siRNA targeted to amyloid-beta precursor protein (APP). It is used for the study of Alzheimer's disease.
More description
|
|
| DC77932 | Mivelsiran sodium |
Mivelsiran sodium is a siRNA targeted to amyloid-beta precursor protein (APP). It is used for the study of Alzheimer's disease.
More description
|
|
| DC77931 | Lufepirsen |
Lufepirsen is an unmodified antisense oligonucleotide targeted to Connexin43 (Cx43). Connexin43 is a specific protein in the eye, which plays a role in wound healing.
More description
|
|
| DC77930 | Lufepirsen sodium |
Lufepirsen sodium is an unmodified antisense oligonucleotide targeted to Connexin43 (Cx43). Connexin43 is a specific protein in the eye, which plays a role in wound healing.
More description
|
|
| DC77929 | Lixadesiran |
Lixadesiran is a siRNA in STP705. It targets to COX-2. STP705 is composed of 2 siRNA oligonucleotides (Pixofisiran and Lixadesiran) that individually target TGF-β1 and COX-2 mRNA.
More description
|
|
| DC77928 | Lixadesiran sodium |
Lixadesiran sodium is a siRNA in STP705. It targets to COX-2. STP705 is composed of 2 siRNA oligonucleotides (Pixofisiran and Lixadesiran) that individually target TGF-β1 and COX-2 mRNA.
More description
|
|
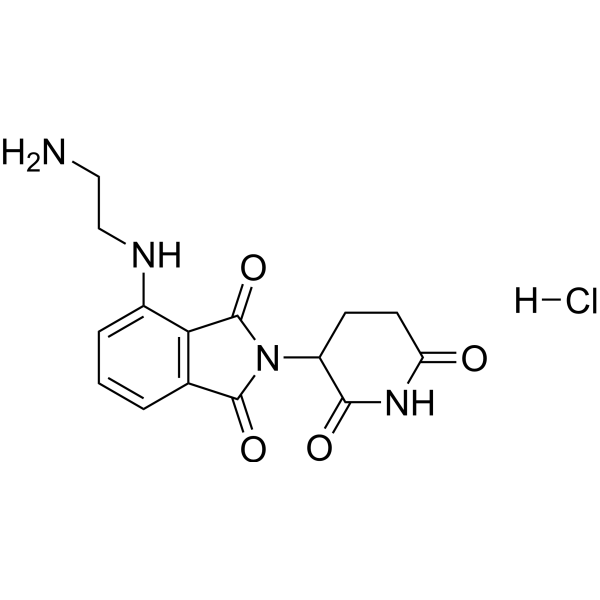
| DC26122 | Pomalidomide-C2-NH2 hydrochloride Featured |
Pomalidomide-C2-NH2 hydrochloride is a synthesized E3 ligase ligand-linker conjugate that incorporates the Pomalidomide based cereblon ligand and a linker used in PROTAC technology.
More description
|
|
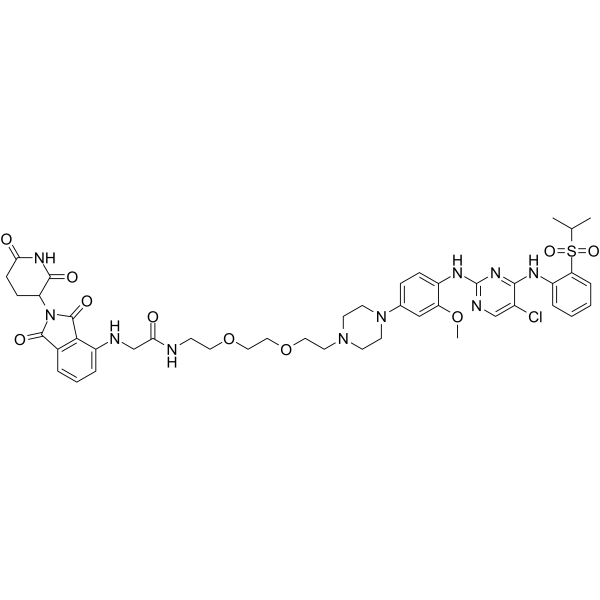
| DC21962 | TL13-12 Featured |
TL13-12 is a novel Anaplastic Lymphoma Kinase (ALK)-PROTAC developed through conjugation of TAE684 and the cereblon ligand pomalidomide.
More description
|
|
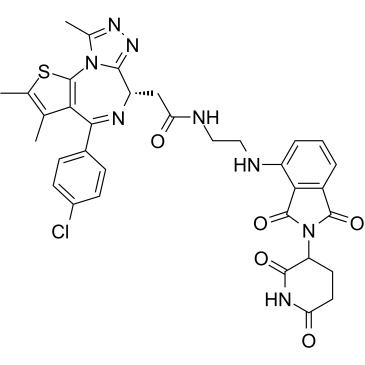
| DC21893 | dBET57 Featured |
dBET57 is a novel BRD4 heterobifunctional small-molecule ligand (PROTAC), exhibits significant and selective degradation of BRD4 BD1 (DC50/5h=500 nM), but is inactive on BRD4 BD2..
More description
|
|
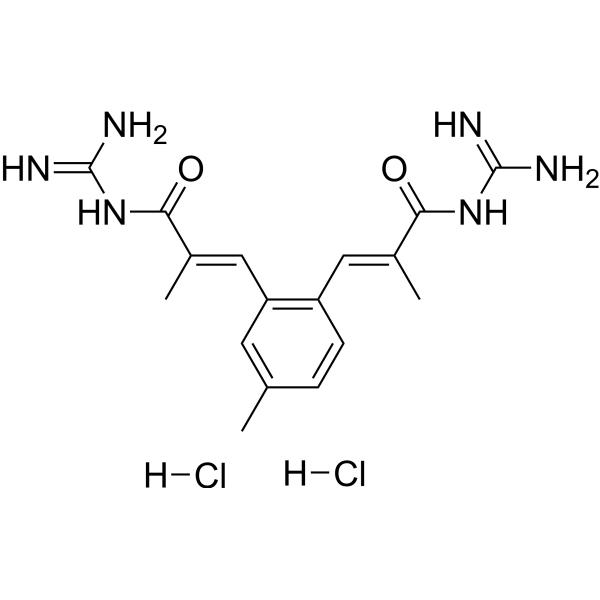
| DC77714 | S3226 Featured |
S3226 is an inhibitor for Na+/H+ exchange subtype 3 (NHE3) with an IC50 of 0.2 µmol/L in rat NHE3 transfected fibroblasts. S3226 exhibits protective activity in rat ischemia-induced acute renal failure models.
More description
|
|
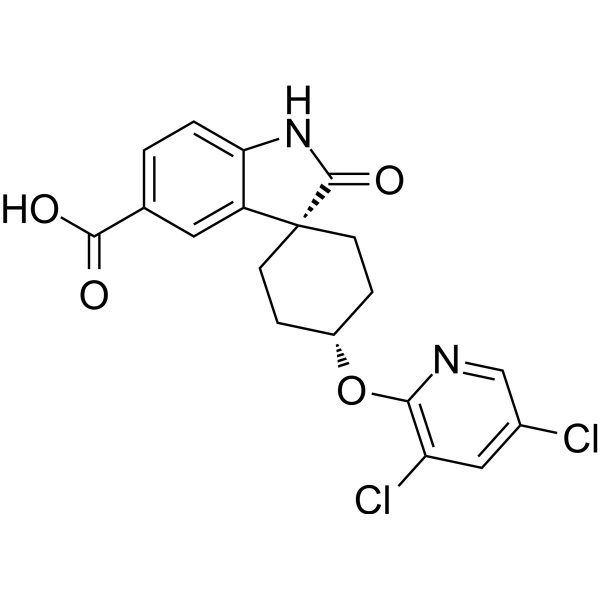
| DC74167 | SC-919 Featured |
SC-919 (SC919) is a potent, selective inhibitor of IP6K (Inositol hexakisphosphate kinase) with IC50 of <5.2, <3.8 and 0.65 nM for human IP6K1/2/3, respectively.
More description
|
|
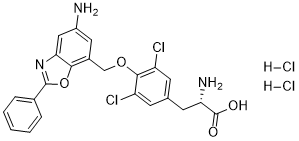
| DC31420 | JPH203 dihydrochloride Featured |
JPH203, also known as KYT-0353, is a potent and selective LAT1 selective ( L-type amino acid transporter 1) inhibitor. JPH203 can very potently inhibit l-leucine uptake. JPH203 inhibits YD-38 cell growth. JPH203 up-regulated the population of apoptotic Y
More description
|
|
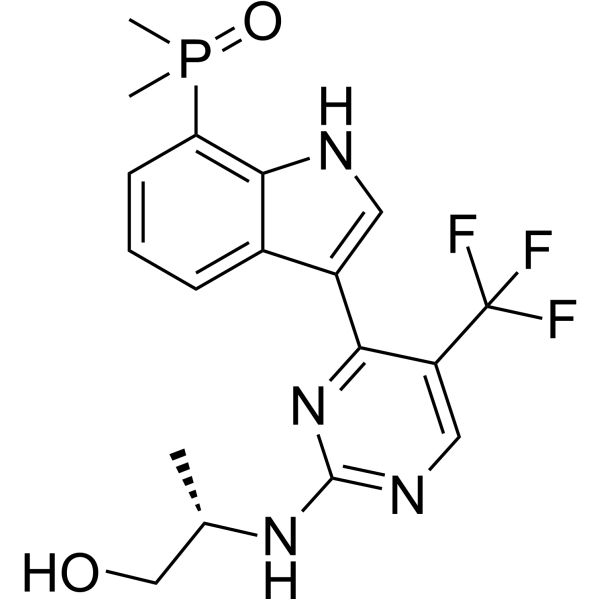
| DC77813 | Zeltociclib Featured |
Zeltociclib is a cyclin-dependent kinase inhibitor with antitumor effects.
More description
|
|
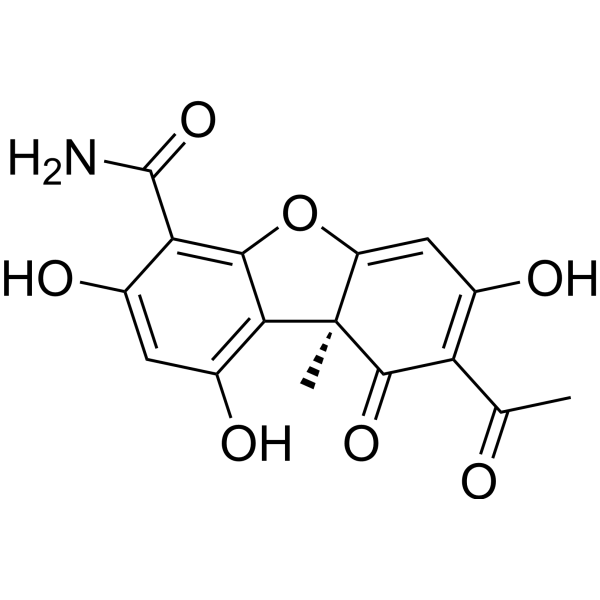
| DC23922 | Cercosporamide Featured |
A broad-spectrum natural antifungal compound that acts as a selective and highly potent fungal Pkc1 kinase inhibitor.
More description
|
|
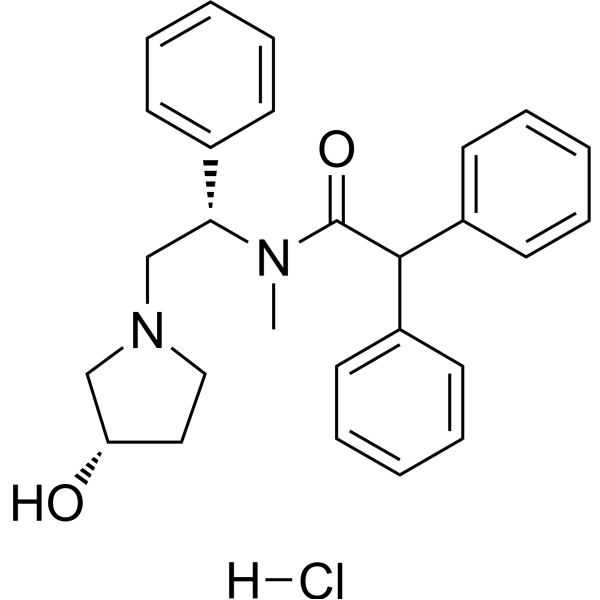
| DC33138 | EMD-61753 hydrochloride Featured |
Asimadoline HCl is a κ-opioid receptor agonist potentially for the treatment of pruritus. Asimadoline has also been shown to be used in the treatment of irritable bowel syndrome.
More description
|
|
| DC7122 | Tazemetostat(EPZ-6438) Featured |
EPZ-6438 is a potent, selective, and orally bioavailable small-molecule inhibitor of EZH2 enzymatic activity with Ki value of 2.5±0.5 nM.
More description
|
|
| DC26003 | TOPK inhibitor-1 HCl (OTS-964 Analogue) Featured |
Novel PDZ binding kinase (PBK) inhibitor.
More description
|
.png)
|
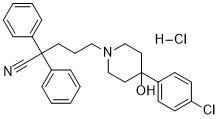
| DC8808 | BX 513 hydrochloride Featured |
BX-513 hydrochloride inhibits MIP-1α-induced intracellular calcium mobilization (IC50 = 2.5 μM).
More description
|
|
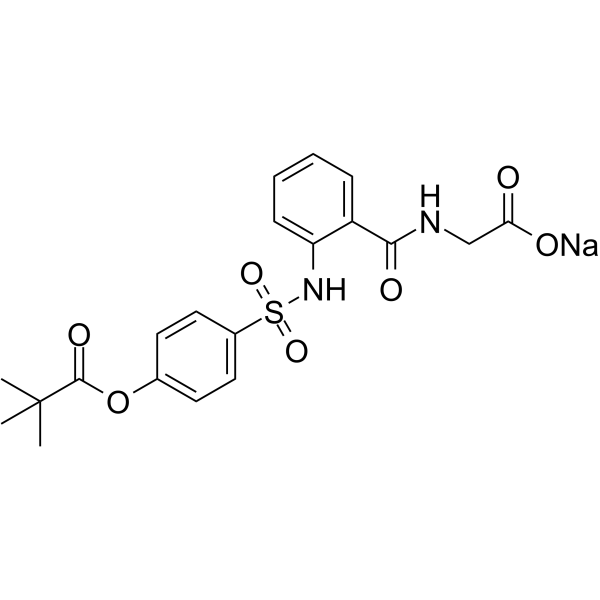
| DC25075 | Sivelestat sodium Featured |
A potent, specific and competitive inhibitor of human neutrophil elastase with IC50 of 44 nM.
More description
|
|
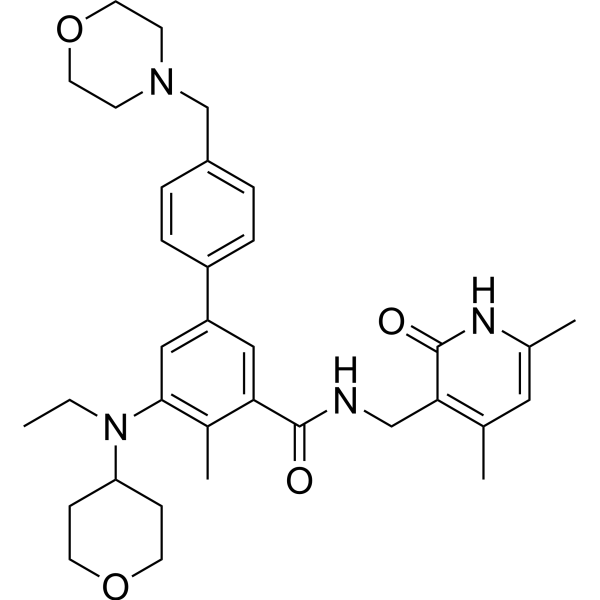
| DC31451 | CCS-1477(CBP-IN-1) Featured |
CCS-1477 is a potent and selective p300/CBP bromodomain inhibitor, is targeted & differentiated from BET inhibitors in prostate cancer cell lines in vitro. Combination of CCS1477 & JQ1 resulted in a highly synergistic inhibitory effect on proliferation in normal 22Rv1 cells. Global gene expression analysis revealed significantly fewer altered genes after CCS1477 (27 up, 119 down) compared to JQ1 (196 up, 655 down).
More description
|
.gif)
|
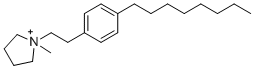
| DC21557 | RB-011 Featured |
A small molecule that disrupt 14-3-3 dimers at low micromolar concentrations and induce rapid down-regulation of Raf-MAPK and PI3K-Akt signaling in Jurkat cells.
More description
|
|
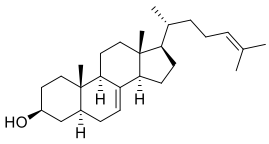
| DC45677 | 5α-Cholesta-7,24-dien-3β-ol Featured |
5α-Cholesta-7,24-dien-3β-ol, a sterol, can be found in hamster cauda epididymal mature spermatozoa.
More description
|
|
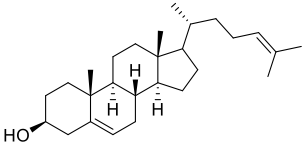
| DC32956 | Desmosterol Featured |
Desmosterol is an endogenous agonist of RORgamma; Intermediate in the synthesis of cholesterol.
More description
|
|
| DC12188 | Lathosterol Featured |
Lathosterol is a cholesterol-like molecule. Serum Lathosterol concentration is an indicator of whole-body cholesterol synthesis.
More description
|

|