 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
| Cat. No. | Product Name | Field of Application | Chemical Structure |
|---|---|---|---|
| DC9383 | Rimonabant (Hydrochloride) Featured |
Rimonabant Hcl(SR141716A) is a selective central cannabinoid (CB1) receptor inverse agonist with Ki of 1.8 nM.
More description
|

|
| DC7721 | RG7090(Basimglurant) Featured |
RG7090(CTEP Derivative,Basimglurant, RO4917523) is a negative modulator of metabotropic glutamate receptor subtype 5 (mGluR5; GRM5).
More description
|

|
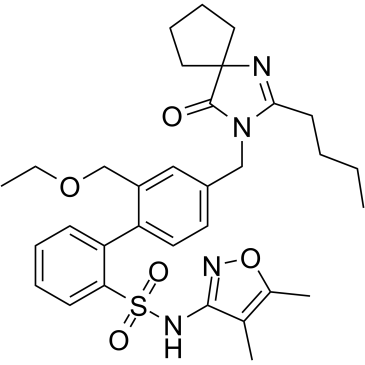
| DC8191 | Sparsentan(PS433540) Featured |
Sparsentan (RE-021) is a highly potent dual angiotensin II and endothelin A receptor antagonist with Kis of 0.8 and 9.3 nM, respectively[1].
More description
|
|
| DC9805 | Preladenant(SCH420814) Featured |
Preladenant(SCH 420814) is a potent and selective antagonist at the adenosine A2A receptor.
More description
|

|
| DC8347 | PRE-084 hydrochloride Featured |
PRE-084 hydrochloride is a high affinity, selective σ1 agonist (Ki values are 2.2 and 13091 nM for σ1 and σ2 receptors respectively).
More description
|

|
| DC7251 | AM 281 Featured |
Potent, selective CB1 cannabinoid receptor antagonist/inverse agonist (Ki values are 12 and 4200 nM for CB1 and CB2 receptors respectively). Increases locomotor activity following systemic administration in vivo. Analog of SR141716A (Ki = 14 nM).
More description
|

|
| DC9742 | Ponesimod Featured |
Ponesimod(ACT-128800) is an orally active, selective sphingosine-1-phosphate receptor 1 (S1P1) immunomodulator with EC50 of 5.7 nM.
More description
|

|
| DC8741 | Plerixafor octahydrochloride Featured |
Plerixafor octahydrochloride(AMD3100 8HCL) is a chemokine receptor antagonist for CXCR4 and CXCL12-mediated chemotaxis with IC50 of 44 nM and 5.7 nM, respectively.
More description
|

|
| DC9465 | Pitolisant (hydrochloride) Featured |
Pitolisant Hcl(BF2.649;Ciproxidine ) is a novel, potent, and selective nonimidazole inverse agonist at the recombinant human H3 receptor (Ki=0.16 nM).
More description
|

|
| DC10012 | Pirmenol hydrochloride Featured |
Pirmenol hydrochloride inhibits IK.ACh by blocking muscarinic receptors. The IC50 of Pirmenol for inhibition of Carbachol-induced IK.ACh is 0.1 μM.
More description
|

|
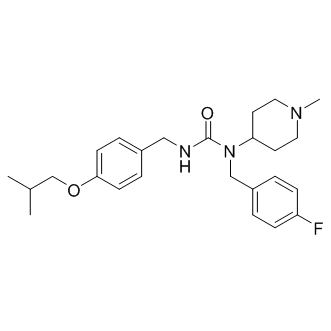
| DC8795 | Pimavanserin(ACP-103) Featured |
Pimavanserin(ACP-103) is a potent and selective 5-HT2A receptor inverse agonist with mean pIC50 of with 8.7 in the cell-based functional assay.
More description
|
|
| DC7480 | PF-3274167(cligosiban) Featured |
PF-3274167 is a high-affinity nonpeptide oxytocin receptor (OTR) antagonist, with Ki of 9.5 nM.
More description
|

|
| DC12316 | PD-168077 maleate Featured |
PD-168077 maleate is a selective dopamine D4 receptor agonist, with a Ki of 9 nM.
More description
|

|
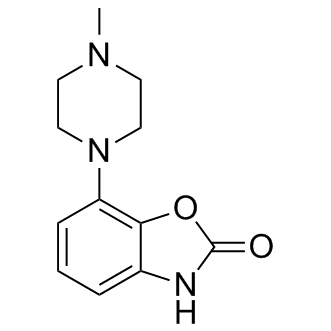
| DC6911 | pardoprunox (SLV308) Featured |
Pardoprunox(SLV-308) is a novel partial dopamine D2 and D3 receptor agonist and serotonin 5-HT1A receptor agonist; D2 (pKi = 8.1) and D3 receptor (pKi = 8.6) partial agonist (IA = 50% and 67%, respectively) and 5-HT1A receptor (pKi = 8.5) full agonist (IA
More description
|
|
| DC8281 | Ozanimod (RPC1063) Featured |
Ozanimod (RPC1063) is a selective S1P1R modulator
More description
|

|
| DC10630 | ONO-7300243 Featured |
ONO-7300243 is a novel, potent LPA1(Lysophosphatidic Acid Receptor) antagonist with an IC50 of 160 nM.
More description
|

|
| DC11043 | Omidenepag Featured |
Omidenepag is a potent, selective agonist human EP2 receptor with binding IC50/EC50 of 10/1.7 nM, >500-fold selectivity over EP1, EP3 and EP4 receptors; Omidenepag is the active form of Omidenepag Isopropyl (OMDI).
More description
|

|
| DC9248 | Olodaterol(BI-1744) hydrochloride Featured |
Olodaterol is a novel, long-acting beta2-adrenergic agonist (LABA) that exerts its pharmacological effect by binding and activating beta2-adrenergic receptors located primarily in the lungs.
More description
|

|
| DC9059 | Olanzapine Featured |
Olanzapine(LY170053) is a high affinity for 5-HT2 serotonin and D2 dopamine receptor antagonist.
More description
|

|
| DC8836 | NIBR189 Featured |
NIBR 189 is a potent and selective EBI2 (GPR183) receptor antagonist (IC50 values are 11 and 15 nM for human and mouse EBI2 receptors respectively).
More description
|

|
| DC10455 | NE-100 Featured |
NE100 hydrochloride is a potent and selective σ1 receptor antagonist (Ki = 0.86 nM) that displays > 55-fold selectivity over σ2 receptors and > 6000-fold selectivity over D1,
More description
|

|
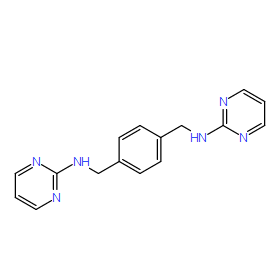
| DC8822 | MSX-122 Featured |
MSX-122 is a n orally bioavailable inhibitor of CXCR4 with potential antineoplastic and antiviral activities.
More description
|
|
| DC10437 | MRE-269 Featured |
MRE-269 is an active metabolite of selexipag, and acts as a selective IP receptor agonist.
More description
|

|
| DC3160 | Montelukast Sodium Featured |
Montelukast belongs to a group of medications known as leukotriene receptor antagonists.
More description
|

|
| DC7781 | ML-224 Featured |
ML-224 (ANTAG3) is a selective TSH receptor antagonist inhibits stimulation of thyroid function in female mice.
More description
|

|
| DC9966 | MK-1064 Featured |
MK-1064 is a selective orexin 2 receptor antagonist (2-SORA) for the research of insomnia.
More description
|

|
| DC10045 | MK-0557 Featured |
MK-0557 is a highly selective, orally administered neuropeptide NPY5R antagonist, could limit weight regain after very-low-calorie diet (VLCD)-induced weight loss.
More description
|

|
| DC9297 | MDK-5220(Orexin-2 receptor agonist) Featured |
MDK-5220(Orexin-2 receptor agonist) is the first selective nonpeptidic orexin 2 receptor (OX2R) agonist (OX2R EC50 = 0.023 μM, Emax = 98%; OX1R EC50 = 1.616 μM, Emax = 100%)
More description
|

|
| DC1012 | Macitentan (Actelion-1,ACT-064992) Featured |
macitentan (Actelion-1, ACT-064992) is an orally active, non-peptide dual endothelin (ET)A/B receptor antagonist with IC50 of 0.5 nM/391 nM.
More description
|
|
| DC7192 | LY-404039 Featured |
LY404039 is an inhibitor for mGluR1(Ki=149 nM) and mGluR2(Ki= 92 nM), which can also inhibit dopamine receptor.
More description
|

|