 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
| Cat. No. | Product Name | Field of Application | Chemical Structure |
|---|---|---|---|
| DC72613 | 3-AQC |
3-AQC, a piperazinylquinoxaline derivative, is a potent and competitive 5-HT3 receptor antagonist.
More description
|

|
| DC72612 | RS-102221 |
RS-102221 is a selective 5-HT2C receptor antagonist (Ki=10 nM). RS-102221 shows nearly 100-fold selectivity for the 5-HT2C receptor as compared to the 5-HT2A and 5-HT2B receptors. RS-102221 can promote the differentiation of new nerve cells. RS-102221 increases food-intake and weight-gain in rats.
More description
|
|
| DC72611 | CP-135807 |
CP-135807 is an orally active and selective 5-HT1D agonist (IC50=3.1 nM), bovine). CP-135807 mediates central psychoactivity and can be used in antidepressant research.
More description
|
|
| DC11437 | VU0652957 Featured |
VU0652957 (VU2957, Valiglurax) is a potent, selective mGlu4 positive allosteric modulator (PAM) with EC50 of 64.6 nM in calcium mobilization human mGlu4/Gqi5 assays; showes excellent pharmacokinetics across species (low CLps, %F > 35%), an acceptable CYP profile (>30 uM vs. 3A4, 2D6 and 2C9, 12.5 uM vs. 2C19 and 1.5 uM vs. 1A2), no CYP induction or timedependent inhibition and excellent metabolite coverage across species; also shows attractive predicted human PK parameters (CLps 5-9 mL/min/kg, Vds 1-2 L/kg and t1/2 2-4 hours).
More description
|

|
| DC11545 | IPAG Featured |
IPAG is a prototypic selective inhibitor of sigma1 receptor that engages the unfolded protein response and induces autophagy in cancer cells.
More description
|

|
| DC7780 | ZM241385 Featured |
ZM 241385 is a Potent A2 adenosine receptor antagonist, with approx 50-fold selectivity for A2A receptors.
More description
|

|
| DC7341 | WZ 811 Featured |
WZ811 is a novel small molecular and potency CXCR4 antagonist with EC50 of 0.3 nM.
More description
|

|
| DC2050 | WIN-55212-2 mesylate Featured |
WIN 55212-2 (mesylate) is a potent aminoalkylindole cannabinoid (CB) receptor agonist with a Ki of 62.3 and 3.3 nM for human recombinant CB1 and CB2 receptors, respectively.
More description
|

|
| DC9255 | VU0357017 Featured |
VU0357017 hydrochloride is a highly selective M1 agonists that appear to act at an allosteric site to activate the receptor (EC50 = 477 ± 172 nM; pEC50 = 6.37 ± 0.15).
More description
|

|
| DC9497 | VU0152100 Featured |
VU0152100 is a potent and selective allosteric potentiator of M4 mAChR with an EC50 of 380 ± 93 nM.
More description
|

|
| DC9928 | VU 0364770 Featured |
VU 0364770 is a positive allosteric modulator(PAM) of mGlu4 with EC50 of 1.1 μM, exhibits insignificant activity at 68 other receptors, including other mGlu subtypes.
More description
|

|
| DC8860 | Vorapaxar Featured |
Vorapaxar (SCH 530348) is a potent and orally active thrombin receptor (PAR-1) antagonist with Ki of 8.1 nM.
More description
|

|
| DC8814 | Vipadenant Featured |
Vipadenant(BIIB-014) is an adenosine A2a antagonist with Ki of 1.3 nM; less potent for A1(Ki=69 nM).
More description
|

|
| DC7336 | Vicriviroc maleate(Sch-417690) Featured |
Vicriviroc Malate is a potent CKR-5 (CCR5) antagonist with IC50 of 0.91 nM.
More description
|

|
| DC9150 | Tianeptine sodium salt Featured |
Tianeptine sodium salt is a selective facilitator of 5-HT uptake in vitro and in vivo.
More description
|

|
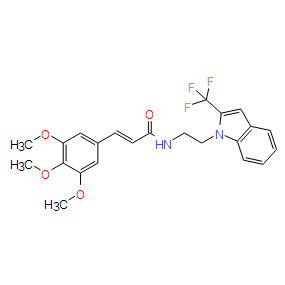
| DC8348 | TG6-10-1 Featured |
TG6-10-1 is a cell-permeable,highly potent, selective, and competitive antagonist of prostaglandin E2 receptor (EP2, Kb = 17.8 nM).
More description
|
|
| DC7513 | TCS 1102 Featured |
TCS 1102 is a potent, dual orexin receptor antagonist (Ki values are 0.2 and 3 nM for OX2 and OX1 receptors respectively).
More description
|

|
| DC5073 | TAK-875(Fasiglifam) Featured |
TAK-875 is a potent, selective and orally bioavailable GPR40 agonist with EC50 of 0.072 μM.
More description
|

|
| DC10664 | Substance P free acid Featured |
Substance P free acid is a Sensory neuropeptide and inflammatory mediator. Exerts excitatory effects on central and peripheral neurons, constricts bronchioles and is involved in pain transmission.
More description
|

|
| DC6906 | Aprepitant (MK-0869, L-754030) Featured |
Substance P antagonists (SPA).
More description
|

|
| DC8614 | Rolapitant Featured |
Rolapitant (SCH619734) is a potent, selective and orally active neurokinin NK1 receptor antagonist with a Ki of 0.66 nM.
More description
|

|
| DC4108 | Sitaxentan sodium Featured |
Sitaxentan sodium (TBC-11251) is a selective endothelin receptor-A antagonist with IC50 and Ki of 1.4 nM and 0.43 nM, respectively.
More description
|

|
| DC8354 | Intepirdine (SB-742457, RVT-101) Featured |
SB-742457(RVT-101) is a selective 5-HT6 receptor antagonist with potential cognition, memory, and learning-enhancing effects.
More description
|
|
| DC9313 | Sarpogrelate (hydrochloride) Featured |
Sarpogrelate(MCI-9042) hydrochloride, a selective 5-HT2 antagonist, has been widely used as an anti-platelet agent for the treatment of PAD.
More description
|

|
| DC9264 | S1RA hydrochloride Featured |
S1RA(E-52862) is a potent and selective sigma-1 receptor(σ1R, Ki=17 nM), showed good selectivity against σ2R (Ki > 1000 nM).
More description
|

|
| DCAPI1593 | Rupatadine Featured |
Rupatadine is a dual histamine H1 and PAF antagonist
More description
|

|
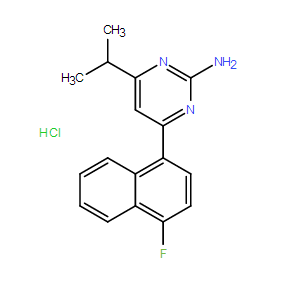
| DC8334 | RS-127445 Featured |
RS-127445 is a potent and selective antagonist at the serotonin 5-HT2B receptor, with around 1000x selectivity over the closely related 5-HT2A and 5-HT2C receptors.
More description
|
|
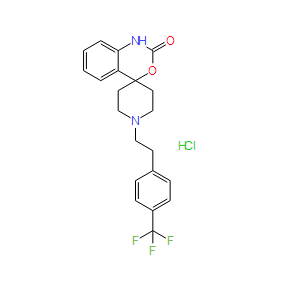
| DC9251 | RS-102895 hydrochloride Featured |
RS 102895 hydrochloride is a chemokine receptor CCR2 antagonist.
More description
|
|
| DC7268 | Risperidone(R 64 766) |
Risperidone(R 64 766) is a serotonin 5-HT2 receptor blocker(Ki= 0.16 nM) and a potent dopamine D2 receptor antagonist(Ki= 1.4 nM).
More description
|

|
| DC8876 | Rimonabant(SR141716) Featured |
Rimonabant(SR141716) is a selective central cannabinoid (CB1) receptor inverse agonist with Ki of 1.8 nM.
More description
|

|