 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
| Cat. No. | Product Name | Field of Application | Chemical Structure |
|---|---|---|---|
| DC72655 | Foslevcromakalim |
Foslevcromakalim (QLS-101) is a ATP-sensitive potassium channel opener. Foslevcromakalim is the prodrug used for ocular hypotensive effect.
More description
|

|
| DC72654 | UB-165 |
UB-165 is a nAChR agonist, being a full agonist of the α3β2 isoform and a partial agonist of the α4β2* isoform, with a Ki value of 0.27 nM for [3H]-nicotine binding in rat brain.
More description
|
|
| DC72653 | NS1219 |
NS1219 ((R)-SPD502) is the isomer of NS 1209. NS1209 is a selective AMPA receptor antagonist with neuroprotective activity. NS1209 can be used for the research of stroke, neuropathic pain and epilepsy.
More description
|
|
| DC72652 | Oleoyl-D-lysine |
Oleoyl-D-lysine is a selective Glycine Transporter-2 (GlyT2) inhibitor based on lipid. Oleoyl-D-lysine reverses neuropathic pain in mice, shows antidrowsiness effect on chronic neuropathic pain. Oleoyl-D-lysine is safe and effective without respiratory depression.
More description
|
|
| DC10493 | WZB117 Featured |
WZB117 is an irreversible inhibitor of glucose transporter 1 (Glut1) that blocks glucose transport in diverse cancer cells (IC50 = ~0.6 µM), reducing extracellular lactate and intracellular ATP levels.
More description
|

|
| DC9193 | Carbamazepine Featured |
Carbamazepine, a sodium channel blocker, is an anticonvulsant drug.
More description
|

|
| DC7100 | CFTRinh 172 Featured |
Voltage-independent, selective CFTR chloride channel blocker (Ki = 300 nM) that alters channel gating.
More description
|

|
| DC8429 | Verdinexor (KPT-335) Featured |
Verdinexor (KPT-335) is an orally bioavailable, selective XPO1/CRM1 inhibitor.
More description
|

|
| DC8916 | Vanoxerine dihydrochloride Featured |
Vanoxerine 2 Hcl(GBR12909) is a potent and selective DRI (Dopamine reuptake inhibitor).
More description
|

|
| DC10619 | UCPH-101 Featured |
UCPH-101 is a selective excitatory amino acid transporter subtype 1 (EAAT1) inhibitor.
More description
|

|
| DC7755 | TRCP6 inhibitor(SAR7334) Featured |
TRPC6 inhibitor is a potent TRPC6(Transient receptor potential cation channel, subfamily C, member 6) inhibitor.
More description
|

|
| DC8896 | Tiagabine hydrochloride Featured |
Tiagabine(NO328) is a selective gamma-aminobutyric acid (GABA) reuptake inhibitor.
More description
|

|
| DC1102 | Tariquidar (XR9576) Featured |
Tariquidar (XR9576) is a potent and selective noncompetitive inhibitor of P-glycoprotein with Kd of 5.1 nM.
More description
|

|
| DC7863 | Talampanel (GYKI 53773) Featured |
Talampanel is a novel anticonvulsant that acts as an allosteric inhibitor of the AMPA receptor.
More description
|

|
| DC9600 | Senicapoc Featured |
Senicapoc(ICA17043) is a potent and selective Gardos channel blocker; blocked Ca(2+)-induced rubidium flux from human RBCs/ inhibited RBC dehydration with IC50s of 11 nM/30 nM, respectively.
IC50 value: 11/30 nM [1]
Target: Gardos channel
in vitro: I
More description
|

|
| DC9635 | SB-705498 Featured |
SB-705498 is a potent, selective and orally bioavailable transient receptor potential vanilloid 1 (TRPV1) receptor antagonist with a pIC50 of 7.1.
More description
|

|
| DC9735 | SB-366791 Featured |
SB-366791 is a selective and competitive VR1 (TRPV1) antagonist that is commonly used in pain research.
More description
|

|
| DC8099 | Saclofen Featured |
Saclofen is a selective GABAB antagonist.
More description
|

|
| DC11325 | RN-1734 Featured |
RN-1734 is a transient receptor potential vanilloid 4 (TRPV4) antagonist, blocking calcium influx induced by the TRPV4 agonist 4α-phorbol 12,13-didecanoate with IC50 values of 2.3, 5.9, and 3.2 µM for human, mouse, and rat TRPV4, respectively.
More description
|

|
| DC10578 | rac BHFF Featured |
Rac BHFF is a potent and selective GABAB receptor positive allosteric modulator that increases the efficacy and potency of GABA ( > 149% and > 15-fold respectively). Orally active and exhibits anxiolytic activity in vivo.
More description
|

|
| DC7483 | PPQ-102 Featured |
PPQ-102 is a potent CFTR inhibitor which can completely inhibited CFTR chloride current with IC 50approximately 90 nM.
More description
|

|
| DC7588 | PNU120596 Featured |
PNU-120596 is a positive allosteric modulator of α7 nAChR with EC50 of 216 nM.
More description
|

|
| DC10113 | Pipequaline Featured |
Pipequaline (PK 8165) is a partial benzodiazepine receptor agonist with anxiolytic activity.
More description
|

|
| DC10834 | PF-06869206 Featured |
PF-06869206 is an Orally Bioavailable Selective Inhibitors of the Sodium-Phosphate Cotransporter NaPi2a (SLC34A1).
More description
|

|
| DC3108 | PF-04971729 (Ertugliflozin) Featured |
PF-04971729 is a potent and selective inhibitor of the sodium-dependent glucose cotransporter 2, is currently in phase 2 trials for the treatment of diabetes mellitus.
More description
|

|
| DC10529 | PF 05089771 Featured |
PF 05089771 is a Nav1.7 channel blocker extracted from patent WO/2010/079443 A1, compound example 788, has an IC50 of 8.6 nM.
More description
|

|
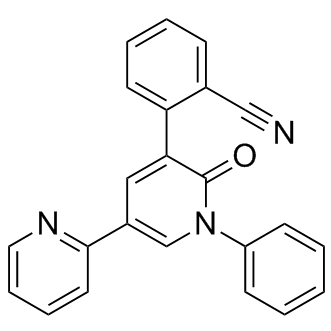
| DC7226 | Perampanel(E2007) Featured |
Perampanel(E2007; ER 155055-90) is a selective noncompetitive AMPA-type glutamate receptor antagonist which has demonstrated anticonvulsant activity in experimental seizure models and antiepileptic activity in clinical trials.
More description
|
|
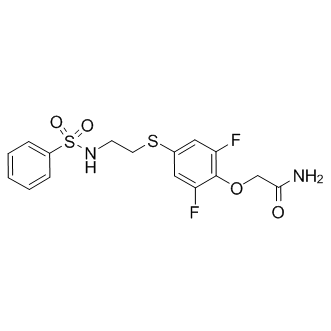
| DC7989 | PEPA Featured |
PEPA is an allosteric modulator of AMPA receptors; binds to the GluA2o and GluA3o LBDs and can be utilized as an indicator of AMPA receptor heterogeneity.
More description
|
|
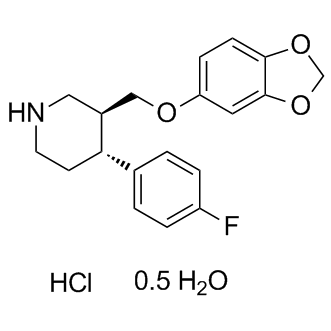
| DC9098 | Paroxetine HCl Featured |
Paroxetine hydrochloride hemihydrate is a antidepressant agents known as selective serotonin-reuptake inhibitors (SSRIs).
More description
|
|
| DC47315 | ML-SI3 Featured |
ML-SI3 is a potent TRPML channel inhibitor with IC50s of 4.7 µM and 1.7 µM for TRPML1 and TRPML2, respectively.
More description
|

|