 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
| Cat. No. | Product Name | Field of Application | Chemical Structure |
|---|---|---|---|
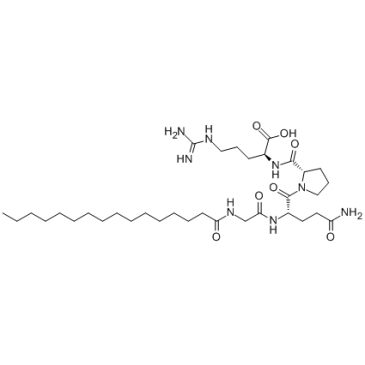
| DC65653 | Palmitoyl tetrapeptide-7 Featured |
Palmitoyl Tetrapeptide-3 (Rigin) is a synthetic peptide, corrspending to 341-344 amino acid sequenceof IgG human H-chain, with phagocytosis stimulating activity.
More description
|
|
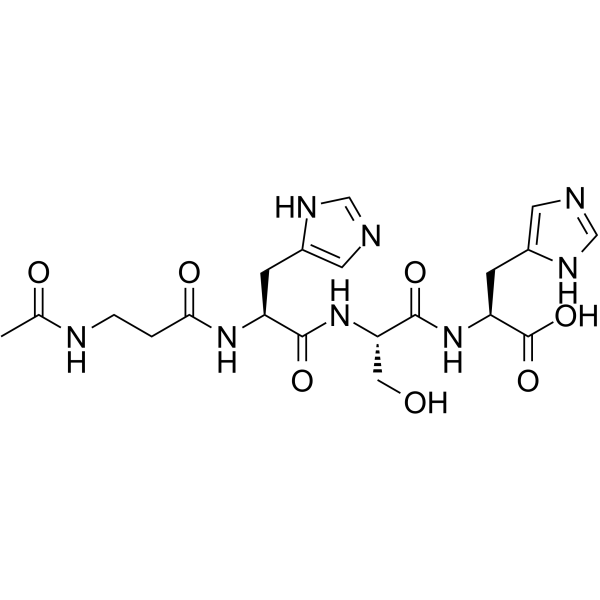
| DC32408 | Ac-beta-ala-his-ser-his-oh Featured |
Eyeseryl, also known as Acetyl Tetrapeptide-5, humectant or hydroscopic moisturizer which helps reduce eye puffiness, improve skin elasticity as well as overall smoothness.
More description
|
|
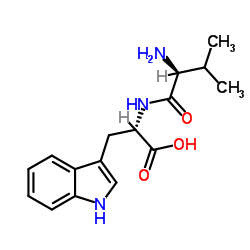
| DC65651 | Dipeptide-2 Featured |
Dipeptide 2 (N-Valyltryptophan; Val-Trp) is a bioactive peptide with anti-aging effect and has been reported used as a cosmetic ingredient.
More description
|
|
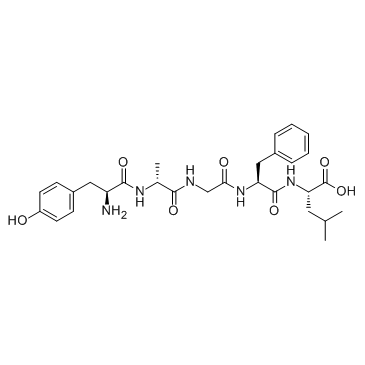
| DC26076 | D-Ala2, D-Leu5-Enkephalin Featured |
An angiotensin II analog that is an agonist of AT1 angiotensin receptor..
More description
|
|
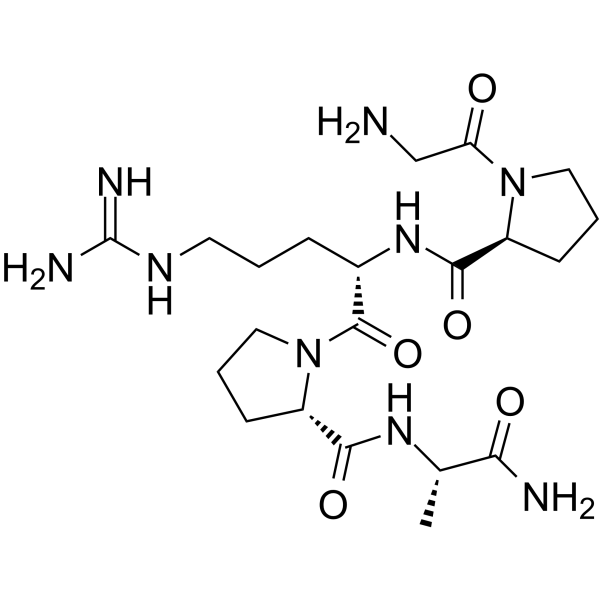
| DC65650 | Vialox Featured |
Pentapeptide-3 is a pentapeptide fragment of neurotoxin waglerin-1 from the venom of Temple Viper. Pentapeptide-3 is a competitive antagonist of nicotinic acetylcholine receptors (nAChRs), it can blocks nerves at the post-synaptic membrane. Pentapeptide-3 has anti-aging effects and it can be used together with other cosmetic peptides.
More description
|
|
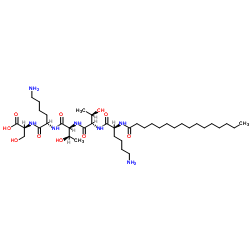
| DC32378 | Palmitoyl pentapeptide Featured |
Matrixyl, also known as Palmitoyl pentapeptide-4 (palmitoyl pentapeptide-3 before 2006) is a matrikine used in anti-wrinkle cosmetics. It was launched in 2000 as an active ingredient for the personal care industry under the trade name Matrixyl by the French cosmetic active ingredient manufacturer Sederma SAS.
More description
|
|
| DC70135 | Bremelanotide Featured |
A peptide, non-selective melanocortin receptor agonist for treatment for female sexual dysfunction; selectively stimulates solicitational behaviors in the female rat, without affecting lordosis, pacing, or other sexual behaviors, does not cause generalized motor activation; a peptide analogue of α-MSH.
More description
|

|
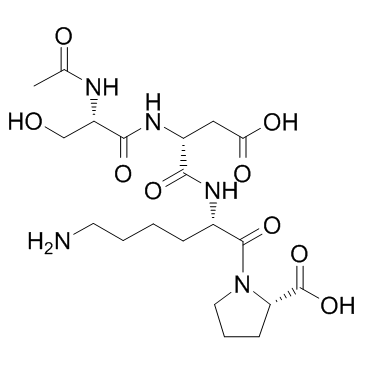
| DC65647 | Goralatide Featured |
N-Acetyl-Ser-Asp-Lys-Pro is a natural and specific substrate for the N-terminal site of ACE.
More description
|
|
| DC32392 | Afamelanotide Featured |
Afamelanotide, also known as CUV1647, is a Melanocortin receptor agonist. Afamelanotide is a synthetic peptide and analogue of α-melanocyte stimulating hormone used to prevent skin damage from the sun in people with erythropoietic protoporphyria .It is under development in other skin disorders in several jurisdictions. It causes skin to turn darker by causing the skin to make more melanin.
More description
|

|
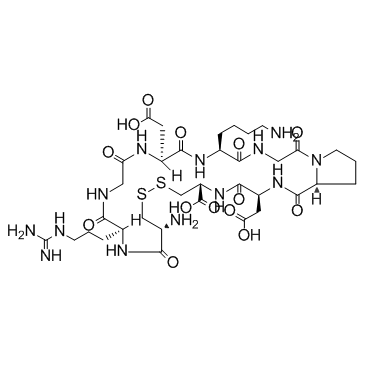
| DC65645 | iRGD Featured |
iRGD peptide is a 9-amino acid cyclic peptide, triggers tissue penetration of drugs by first binding to av integrins, then proteolytically cleaved in the tumor to produce CRGDK/R to interact with neuropilin-1, and has tumor-targeting and tumor-penetrating properties.
More description
|
|
| DC65644 | Nafarelin Featured |
Nafarelin is a gonadotropin-releasing hormone (GnRH) agonist that stimulates secretion of luteinizing hormone (LH) and follicle-stimulating hormone (FSH). Sequence: {Glp}-His-Trp-Ser-Tyr-{2-Naph-Ala}-Leu-Arg-Pro-Gly-NH2.
More description
|

|
| DC65642 | Thymosin beta 4 Featured |
Thymosin beta 4 is a potent regulator of actin polymerization in living cells. Sequence: Ser-Asp-Lys-Pro-Asp-Met-Ala-Glu-Ile-Glu-Lys-Phe-Asp-Lys-Ser-Lys-Leu-Lys-Lys-Thr-Glu-Thr-Gln-Glu-Lys-Asn-Pro-Leu-Pro-Ser-Lys-Glu-Thr-Ile-Glu-Gln-Glu-Lys-Gln-Ala-Gly-Glu-Ser.
More description
|

|
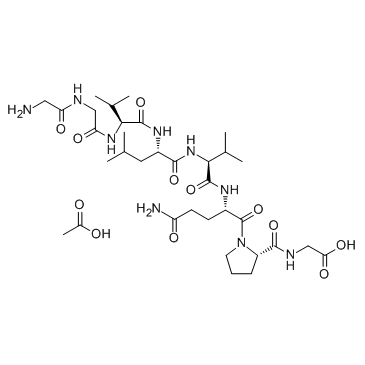
| DC65641 | Larazotide Acetate Featured |
Larazotide acetate is a synthetic peptide that functions as a tight junction regulator and reverses leaky junctions to their normally closed state.
More description
|
|
| DC47396 | Abaloparatide (BA058) Featured |
Abaloparatide (BA058, BIM-44058, ITM-058) is a novel 34-amino acid peptide selected to be a potent and selective activator of the parathyroid hormone receptor (PTH1R) signaling pathway with an IC50 of 0.117 nM in SOST analysis.
More description
|

|
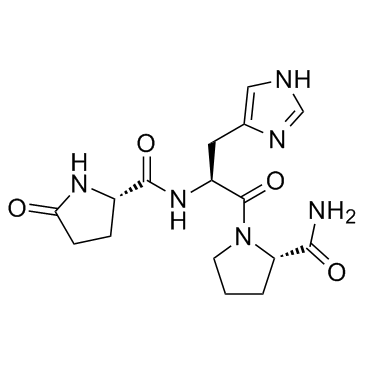
| DC10241 | Protirelin Featured |
Protirelin is a tripeptide that stimulates the release of thyrotropin and prolactin.
More description
|
|
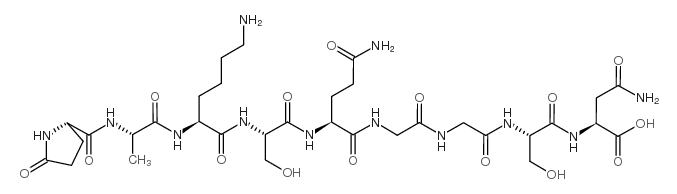
| DC65639 | Thymulin Acetate Featured |
Serum thymic factor (Thymulin) is a zinc-dependent immunomodulatory peptide. Serum thymic factor induces hyperalgesia. Serum thymic factor protects rats from Cephaloridine (HY-B2072)-induced nephrotoxicity by inhibiting ERK activation. Serum thymic factor has anti-diabetic, analgesic and anti-inflammatory effects.
More description
|
|
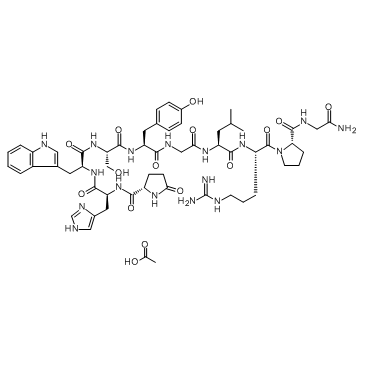
| DC32017 | AY-24031 Featured |
Gonadorelin is a decapeptide that stimulates the synthesis and secretion of both pituitary gonadotropins, LUTEINIZING HORMONE and FOLLICLE STIMULATING HORMONE. GnRH is produced by neurons in the septum PREOPTIC AREA of the HYPOTHALAMUS and released into the pituitary portal blood, leading to stim. Gonadorelin has been shown to have gonadotropin-releasing effects upon the anterior pituitary.
More description
|
|
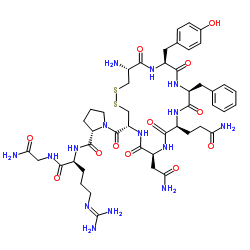
| DC32011 | Argpressin Acetate Featured |
Argpressin is the predominant form of mammalian antidiuretic hormone. Argpressin is a nonapeptide containing an ARGININE at residue 8 and two disulfide-linked CYSTEINES at residues of 1 and 6. Arg-vasopressin is used to treat diabetes insipidus or to improve vasomotor tone and blood pressure.
More description
|
|
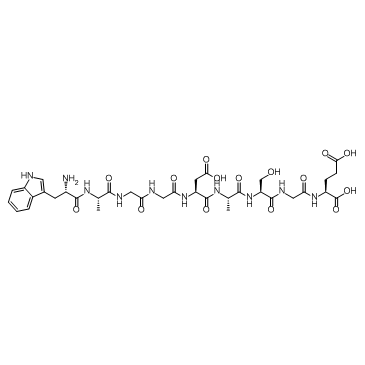
| DC65638 | DSIP Featured |
Delta-Sleep Inducing Peptide trifluoroacetate salt is a neuropeptide, with antioxidant and anxiolytic properties.
More description
|
|
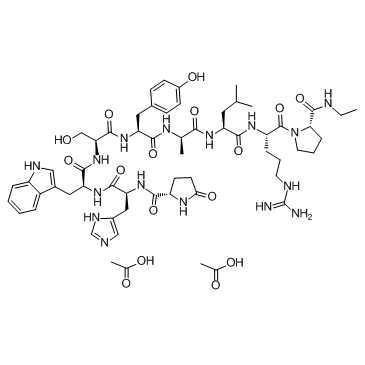
| DC32010 | Alarelin Acetate Featured |
Alarelin is a potent LH-RH agonist in rats and mice. Alarelin reversibly delays sexual maturation in rats, stimulates spawning activity in fish. GnRH ( gonadotropin-releasing hormone, Glp-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly-NH2), which is also referred as LHRH (luteinizing hormone-releasing hormone) or gonadorelin, is crucial for mammalian reproduction and is released from hypothalamic neurons. It is responsible for the secretion of gonadotropins, luteinizing hormone (LH) and follicle-stimulating hormone (FSH), from the pituitary glands.
More description
|
|
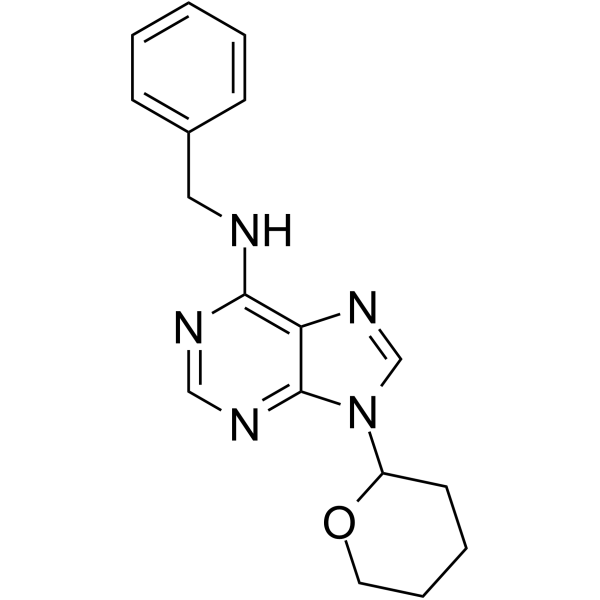
| DC42362 | BAP9THP Featured |
BAP9THP is a synthetic cytokinin derivative and a growth regulator. BAP9THP promotes chlorophyll retention (and senescence delay) in plant tissues exceptionally strongly, and growth of tobacco callus almost as strongly as 6-Benzylaminopurine (BAP). BAP9THP induces adventitious shoot formation ignificantly more strongly than N6-isopentenyladenine or Kinetin.
More description
|
|
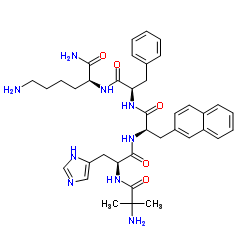
| DC32133 | Ipamorelin Featured |
Ipamorelin, also known as NNC-26-0161, is a a ghrelin mimetic. Ipamorelin counteracts glucocorticoid-induced decrease in bone formation of adult rats. Ipamorelin may ameliorate the symptoms in patients with POI. Ipamorelin accelerates gastric emptying in a rodent model of postoperative ileus through the stimulation of gastric contractility by activating a ghrelin receptor-mediated mechanism involving cholinergic excitatory neurons.
More description
|
|
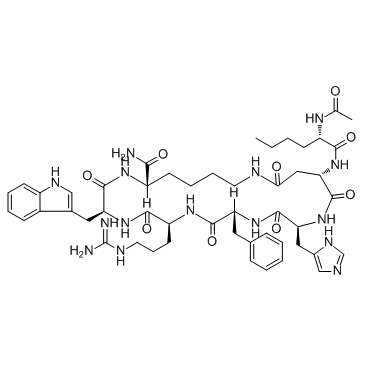
| DC65630 | Melanotan II Featured |
Melanotan (MT)-II, a synthetic melanocortin receptor agonist, is an injectable peptide hormone used to promote tanning.
More description
|
|
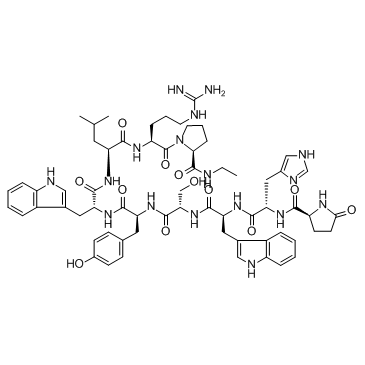
| DC65629 | Deslorelin Featured |
Deslorelin is a GnRH agonist. Deslorelin implants can be used as an alternative to dopamine agonists to induce fertil eoestrus in the bitch in anoestrus. The deslorelin implant can be used successfully in the queen for oestrus inhibition. Deslorelin acetate did not significantly affect the spontaneous contraction amplitude but caused a decrease in the frequency in the dorsal and ventral parts of the bladder.
More description
|
|
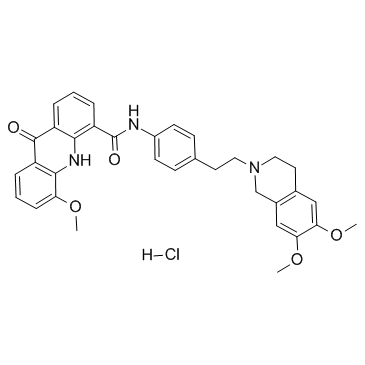
| DC22621 | Elacridar hydrochloride Featured |
Elacridar Hcl (GF120918; GW0918) is a P-glycoprotein inhibitor, and has been used both in vitro and in vivo as a tool inhibitor of P-glycoprotein (Pgp) to investigate the role of transporters in the disposition of various test molecules.IC50 value:Target: P-glycoprotein In vitro, GF120918A demonstrated high plasma protein binding across species, although a definitive protein binding evaluation was precluded by poor recovery, particularly in buffer and in mouse, rat, and dog plasma. GF120918A did not demonstrate potent inhibition of several human cytochrome P450 enzymes evaluated in vitro, with IC(50) values well above concentrations anticipated to be achieved in vivo. Together, these data confirm the utility of GF120918A as a tool P-glycoprotein inhibitor in preclinical species and offer additional guidance on preclinical dose regimens likely to produce P-glycoprotein-mediated effects.
More description
|
|
| DC10957 | MB725 Featured |
MB725 is a small-molecule p53 mutant Y220C stabilizer, induces selective viability reduction in several p53-Y220C cancer cell lines (Huh7 cell IC50=10 uM).
More description
|

|
| DC53049 | CVN424 Featured |
CVN424 is a potent and selective GPR6 inverse agonist (EC50 of 38 nM) for the treatment of parkinson's disease.
More description
|
|
| DC9755 | eFT508(Tomivosertib) Featured |
Tomivosertib (eFT-508) is a potent and selective MNK1/2 inhibitor with IC50s of 2.4 nM and 1 nM, respectively. It potentially results in decreased tumor cell proliferation and tumor growth. Tomivosertib (eFT-508) inhibits eIF4E phosphorylation and dramatically downregulates PD-L1 protein abundance.
More description
|

|
| DC41888 | WL47 Featured |
WL47 is a selective, high-affinity, disrupter of cavolin-1 oligomers (Kd=23 nM) than BSA, casein, and HEWL. WL47 is a caveolin-1 (CAV1) ligand and is 80% smaller in length than the original T20 parent sequence. WL47 can be used for the study of caveolin-1 function.
More description
|

|
| DC42397 | WL47 TFA Featured |
WL47 TFA is a selective, high-affinity, disrupter of cavolin-1 oligomers (Kd=23 nM) than BSA, casein, and HEWL. WL47 TFA is a caveolin-1 (CAV1) ligand and is 80% smaller in length than the original T20 parent sequence. WL47 TFA can be used for the study of caveolin-1 function.
More description
|
|