 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
| Cat. No. | Product Name | Field of Application | Chemical Structure |
|---|---|---|---|
| DC11589 | dBRD9 Featured |
dBRD9 is a PROTAC that bridge the BRD9 bromodomain and the cereblon E3 ubiquitin ligase complex.
More description
|

|
| DC71542 | 2-MPPA Featured |
2-MPPA (GPI-5693) is an orally active and selective glutamate carboxypeptidase II (GCP II; PSMA) inhibitor with an IC50 of 90 nM.
More description
|

|
| DC7495 | SB-334867 HCl Featured |
SB-334867 is a selective non-peptide orexin OX1 receptor antagonist with a pKb value of 7.2.
More description
|

|
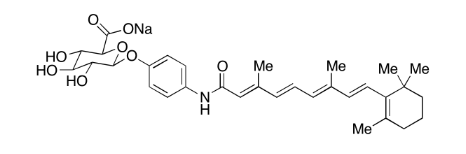
| DC65676 | Fenretinide Glucuronide Monosodium Salt Featured |
Fenretinide Glucuronide Monosodium Salt, is the metabolite of Fenretinide (F250000), which is a synthetic retinoid deriverative, substances related to vitamin A. They are also shown to be used for the treatment of cancer, as well as in the treatment of cystic fibrosis, rheumatoid arthritis, acne, and psoriasis.
More description
|
|
| DC70246 | BFL-1 inhibitor 4E14 Featured |
BFL-1 inhibitor 4E14 (4E14) is a potent, selective, covalent BFL-1 inhibitor that disrupt BH3-binding activity with IC50 of 1.3 uM, targets a unique C55 residue in the BFL-1 groove.4E14 blocks BFL-1 suppression of BAX-mediated mitochondrial apoptosis.
More description
|

|
| DC11818 | SCB-4380 Featured |
SCB-4380 (SCB 4380, SCB4380) is the first potent, selective inhibitor for PTPRZ (protein tyrosine phosphatase receptor-type Z) with IC50 of 0.4 uM against human Z-ICR-catalyzed hydrolysis.
SCB-4380 also strongly inhibits PTPRG (IC50=0.4 uM), and shows little inhibition on PTPRA, PTPRM, PTPRS, PTPRB, PTPN1 and PTPN6.
SCB-4380 inhibites PTPRZ activity in C6 glioblastoma cells, and suppresse cell migration and proliferation in vitro and tumor growth in a rat allograft model.
More description
|

|
| DC7709 | ATN-161 Featured |
ATN-161 is a novel integrin α5β1 antagonist, which inhibits angiogenesis and growth of liver metastases in a murine model.
More description
|
|
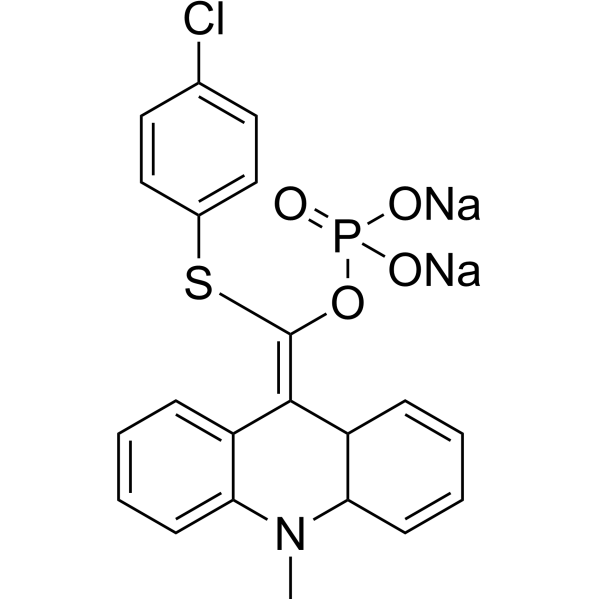
| DC65677 | Lumigen APS-5 Featured |
Lumigen APS-5 is a substrate of alkaline phosphatase (ALP). Lumigen APS-5 can be used to assess the activity of alkaline phosphatase (ALP).
More description
|
|
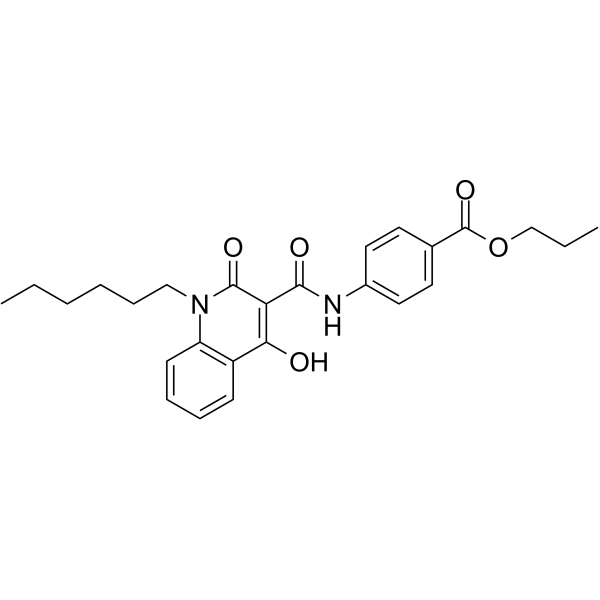
| DC23876 | GSA-10 Featured |
GSA-10 is a novel small-molecule positive modulator of Smoothened with EC50 of 1.2 uM in the differentiation assay.
More description
|
|
| DC65675 | 4-Methylindoline Featured |
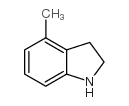
|
|
| DC33707 | Valinomycin Featured |
Valinomycin is an cyclododecadepsipeptide ionophore antibiotic that acts as an insecticide and nematocide.
More description
|

|
| DC22403 | Casopitant mesylate Featured |
Casopitant (GW679769) is a potent, centrally-acting neurokinin 1 (NK1) receptor antagonist (pKi=9.48) with antidepressant and antiemetic activities.
More description
|
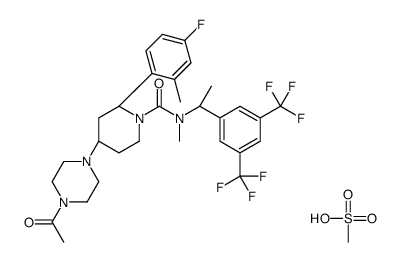
|
| DC4132 | CP-547632 Featured |
CP-547632 is as a potent inhibitor of the VEGFR-2 and basic fibroblast growth factor (FGF) kinases (IC50 11 and 9 nM, respectively).
More description
|

|
| DC32961 | PHA-543613 Featured |
PHA-543613 acts as a potent and selective agonist for the α7 subtype of neural nicotinic acetylcholine receptors, with a high level of brain penetration and good oral bioavailability. It is under development as a possible treatment for cognitive deficits in schizophrenia.
More description
|
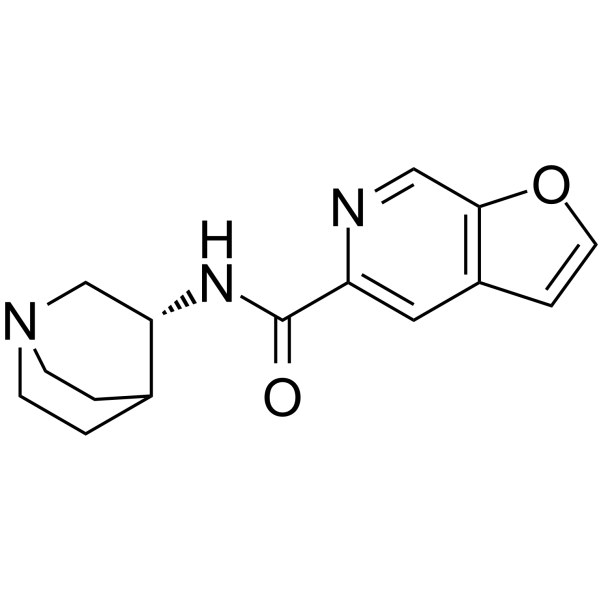
|
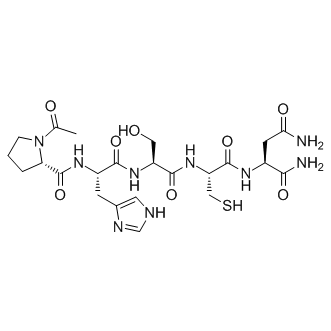
| DC65673 | PAD-IN-2 Featured |
PAD-IN-2 is a potent pad4 inhibitor (IC50: <1 μM). PAD-IN-2 can be used in the research of auto-immune diseases and cancers, such as rheumatoid arthritis, vasculitis, systemic lupus erythematosis, cutaneous lupus erythematosis, ulcerative colitis, cystic fibrosis, asthma, multiple sclerosis and psoriasis.
More description
|
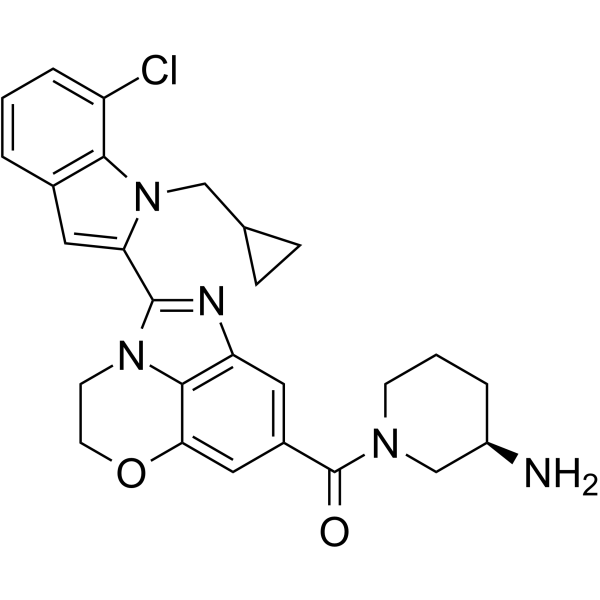
|
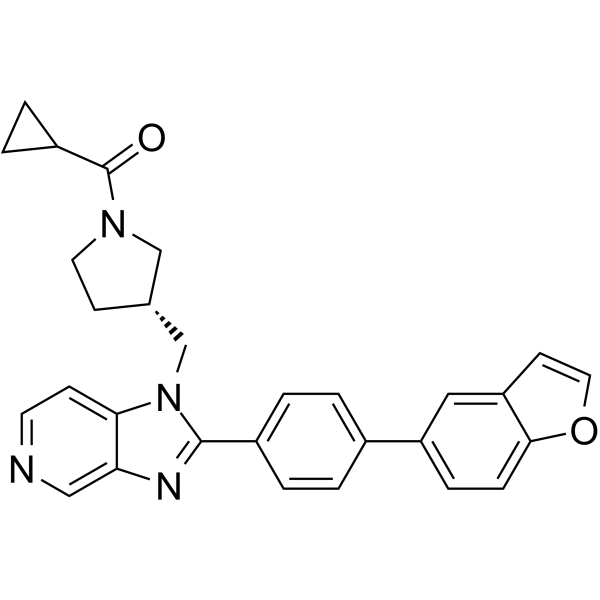
| DC65672 | FASN-IN-5 Featured |
FASN-IN-5 (example 11), a FASN inhibitor, can be used for the research of TH17- or CSF1 -mediated disease or disorder such as cancer, immunological disorders, and obesity.
More description
|
|
| DC9325 | Cilazapril (monohydrate) Featured |
Cilazapril Monohydrate is a angiotensin-converting enzyme (ACE) inhibitor used for the treatment of hypertension and congestive heart failure.
More description
|

|
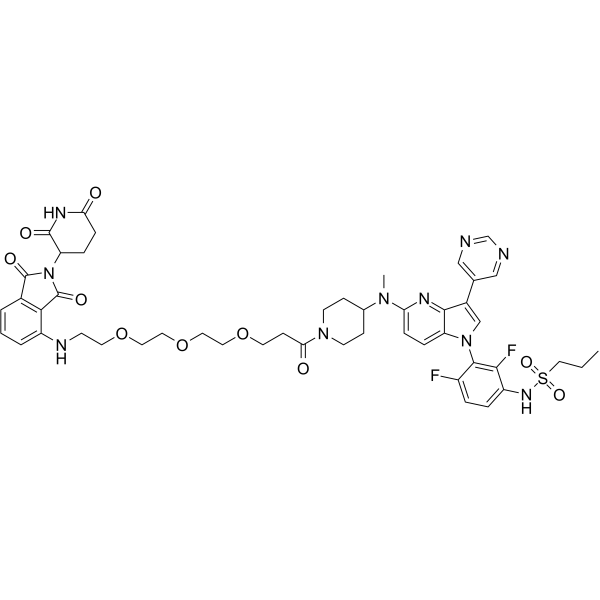
| DC65671 | PROTAC BRAF-V600E degrader-1 Featured |
PROTAC BRAF-V600E degrader-1 is a potent PROTAC BRAF-V600E degrader with Kd value of 2.4 nM and 2 nM for BRAF and BRAF-V600E, respectively. PROTAC BRAF-V600E degrader-1 degrades BRAF-V600E via the ubiquitin-proteasome system (UPS). PROTAC BRAF-V600E degrader-1 can inhibit melanoma cell growth.
More description
|
|
| DC7580 | Naxagolide Featured |
For the detailed information of Naxagolide, the solubility of Naxagolide in water, the solubility of Naxagolide in DMSO, the solubility of Naxagolide in PBS buffer, the animal experiment (test) of Naxagolide, the cell expriment (test) of Naxagolide, the in vivo, in vitro and clinical trial test of Naxagolide, the EC50, IC50,and Affinity of Naxagolide, Please contact DC Chemicals..
More description
|

|
| DC45710 | Pomalidomide-amino-PEG5-NH2 hydrochloride Featured |
Pomalidomide-amino-PEG5-NH2 hydrochloride is a synthesized E3 ligase ligand-linker conjugate that incorporates the Pomalidomide based cereblon ligand and a linker used in PROTAC technology.
More description
|

|
| DC45712 | Pomalidomide-amino-PEG3-NH2 hydrochloride Featured |
Pomalidomide-amino-PEG3-NH2 hydrochloride is a synthesized E3 ligase ligand-linker conjugate that incorporates the Pomalidomide based cereblon ligand and a linker used in PROTAC technology.
More description
|

|
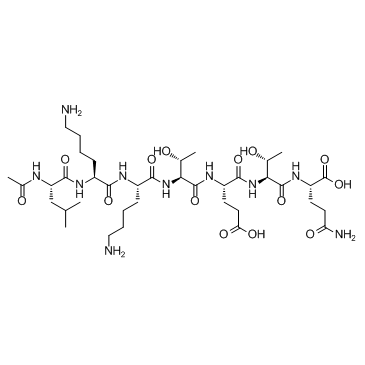
| DC65669 | TB500 Featured |
TB500 is a synthetic version of an active region of thymosin β4. TB500 is claimed to promote endothelial cell differentiation, angiogenesis in dermal tissues, keratinocyte migration, collagen deposition and decrease inflammation.
More description
|
|
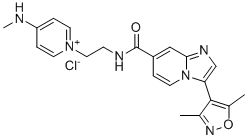
| DC65221 | BAY-6096 Featured |
BAY6096, a potent, selective, and highly water-soluble α2B antagonist with IC50 = 14 nM.
More description
|
|
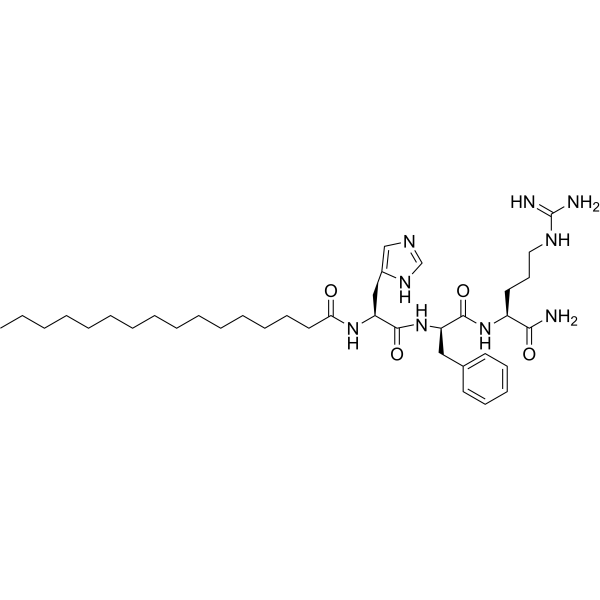
| DC65665 | Palmitoyl Tripeptide-8 Featured |
Palmitoyl tripeptide-8 s a bioactive peptide with anti-allergen effect and has been reported used as a cosmetic ingredient.
More description
|
|
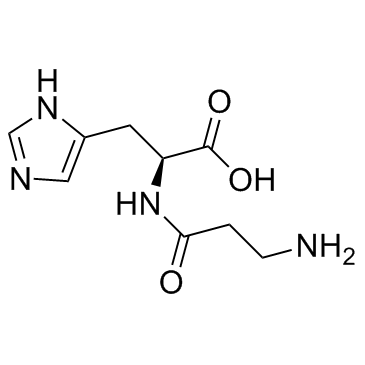
| DC65662 | L-carnosine Featured |
L-Carnosine is a dipeptide of the amino acids beta-alanine and histidine and has the potential to suppress many of the biochemical changes that accompany aging.
More description
|
|
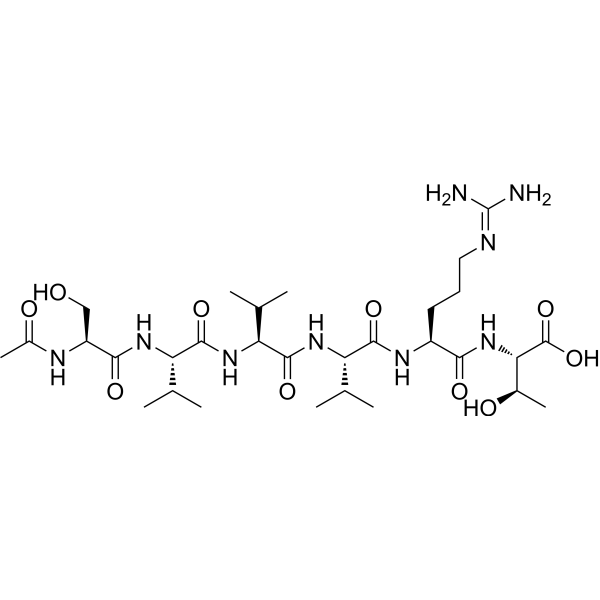
| DC65661 | Acetyl hexapeptide 38 Featured |
Acetyl hexapeptide-38 is a bioactive peptide with upregulate adipogenesis effect and has been reported used as a cosmetic ingredient.
More description
|
|
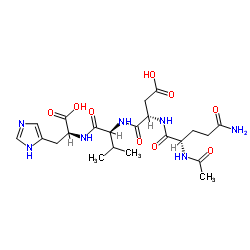
| DC65659 | Acetyl tetrapeptide-9 Featured |
Caprooyl-tetrapeptide-9 (AcTP1) is a bioactive peptide with anti-aging effect and has been reported used as a cosmetic ingredient.
More description
|
|
| DC33607 | Myristoyl Pentapeptide-17 Featured |
Myristoyl Pentapeptide-17 is small peptide. Myristoyl Pentapeptide-17 promotes the delivery of key ingredients for quicker lash growth, thus stimulates the hair growth at the follicle. Myristoyl Pentapeptide-17 effectively promotes the growth of eyelashes. It lengthens and thickens the eyelashes, fortifies the hair and noticeably intensifies eye expression.
More description
|

|
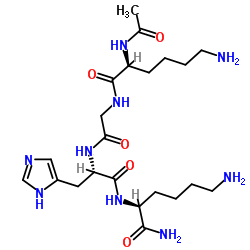
| DC65656 | Acetyl tetrapeptide-3 Featured |
Acetyl tetrapeptide-3 is a bioactive chemical.
More description
|
|
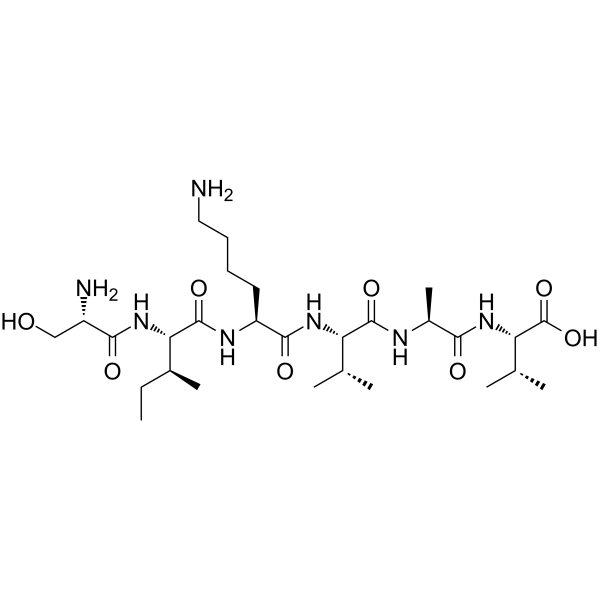
| DC65654 | Hexapeptide-10 Featured |
Hexapeptide-10 is an amino acid derivative.
More description
|
|