 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
| Cat. No. | Product Name | Field of Application | Chemical Structure |
|---|---|---|---|
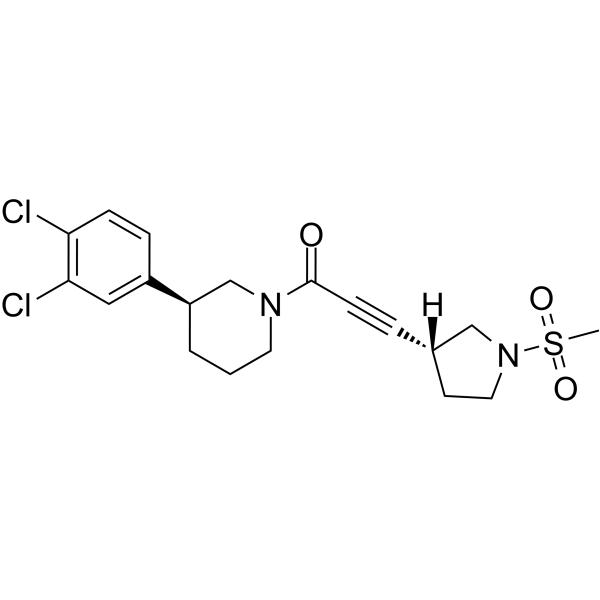
| DC72043 | VVD-118313 Featured |
VVD-118313 (compound 5a) is a potent, selective JAK1 inhibitor. VVD-118313 targets an isoform-restricted allosteric cysteine to block JAK1-dependent trans-phosphorylation and cytokine signaling. VVD-118313 can be used for research of cancer.
More description
|
|
| DC44102 | IPN60090 |
IPN-60090 is an orally active and highly selective inhibitor of glutaminase 1 (GLS1; IC50=31 nM), with no activity observed against GLS-2. IPN-60090 exhibits excellent physicochemical and pharmacokinetic properties in vivo. IPN-60090 can be used for solid tumors research, such as lung and ovarian cancers.
More description
|

|
| DC70372 | DX308 Featured |
DX-308 is a potent, selective dual CYP26A1/B1 inhibitor and retinoic acid metabolism blocking agent.DX308 does not interact with off‐target nuclear receptors or CYP450s, is not genotoxic, and is stable in skin, despite vigorous hepatic metabolism.Topical DX308 induces comedolysis and epidermal thickening without apparent adverse effects in vivo.DX308 shows potent modulation of retinoid‐responsive genes by DX308 in both healthy and keratinization disorder keratinocytes (KCs).DX 308 may present an improved therapeutic alternative for the treatment of keratinization disorders and other retinoid‐responsive skin ailments.
More description
|

|
| DC70373 | DX314 Featured |
DX314 is a potent, specific CYP26B1 inhibitor with IC50 of 108 nM, >15-fold selectivity over CYP26A1.DX314 potentiates all-trans-RA (atRA) gene expression effects in healthy and diseased reconstructed human epidermis (RHE).DX314 potentiates the effects of atRA on the expression and localization of keratin 10 (KRT10), protects barrier function in RHE.DX314 reduces comedonal number, induces epidermal thickening, and increases comedonal profile, while having no effect on transepidermal water loss (TEWL) in treated rhino mice.
More description
|

|
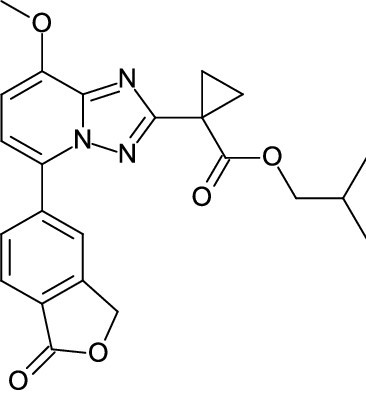
| DC39261 | LEO 39652 Featured |
LEO 39652 is a novel PDE4 inhibitor with IC50 of 3.8 nM and selected as a clinical candidate as it is potent and rapidly degraded by blood and liver to inactive metabolites.
More description
|
|
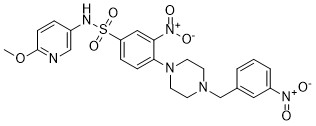
| DC65508 | MyD88-IN-1 Featured |
MyD88-IN-1 is a potent MyD88 inhibitor.
More description
|
|
| DC45287 | TRPC6-PAM-C20 Featured |
TRPC6-PAM-C20 is a selective TRPC6 positive allosteric modulator. TRPC6-PAM-C20 selectively activates TRPC6 over other TRP channels and also activates TRPA1. TRPC6-PAM-C20 induces transient increase in intracellular Ca2+ in HEK cells expressing TRPC6 (EC50=2.37 μM).
More description
|

|
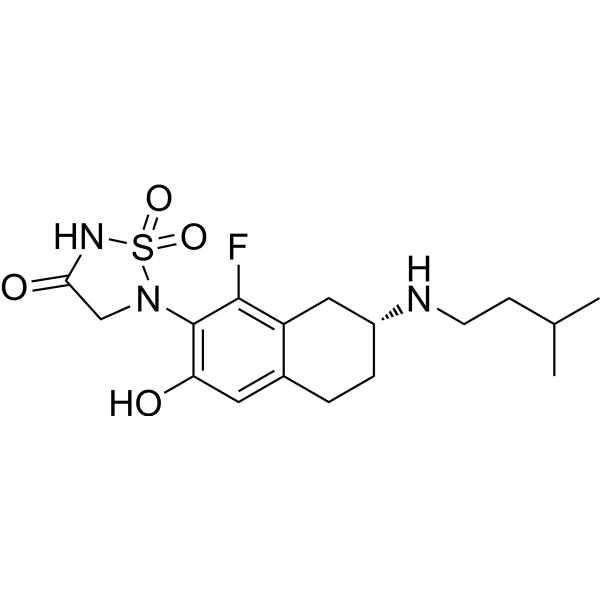
| DC65567 | ABBV-CLS-484 Featured |
Osunprotafib (ABBV-CLS-484) is a potent PTPN1 and PTPN2 inhibitor with subnanomolar activity. Osunprotafib has antitumor activity, enhances the immune response and increases the sensitivity of tumors to immune-mediated killing.
More description
|
|
| DC41049 | Trotabresib (CC-90010) Featured |
Trotabresib (CC-90010) is a reversible and orally active BET inhibitor. CC-90010 is applied in the study for advanced solid tumors.
More description
|

|
| DC44111 | Ibezapolstat |
Ibezapolstat (ACX-362E) is a first-in-class, orally active DNA polymerase IIIC (pol IIIC) inhibitor, with a Ki of 0.325 μM for the DNA pol IIIC from C. difficile. Ibezapolstat is developed for the research of C. difficile infection(CDI).
More description
|

|
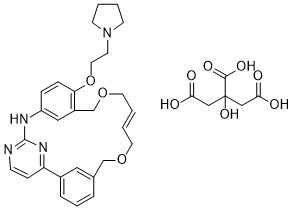
| DC89083 | Pacritinib citrate Featured |
Pacritinib Citrate is the citrate salt form of pacritinib, an orally bioavailable inhibitor of Janus kinase 2 (JAK2).
More description
|
|
| DC22728 | M-110 |
M-110 is a potent, selective inhibitor of PIM kinase family with preference for PIM-3 with IC50 of 0.047, 2.5 and 2.5 uM for PIM3, PIM1 and PIM2, respectively.
More description
|

|
| DC70788 | SMN2 splicing modulator TEC-1 Featured |
SMN2 splicing modulator TEC-1 is a novel specific, CNS penetrant small molecule SMN2 splicing modulator, increases the expression level of FL-SMN2 mRNA and decreases the expression level of Δ7 mRNA.TEC-1 showed higher selectively (>60-fold) on galactosylceramidase and huntingtin gene expression compared to previously reported compounds (e.g., SMN-C3) due to off-target effects on cryptic exon inclusion and nonsense-mediated mRNA decay.TEC-1 modulates SMN2 splicing and displays disease-modifying effects in motor neurons derived from SMA patient iPSCs (GM24468).|TEC-1 rescues the phenotype in a murine model of spinal muscular atrophy (SMA).
More description
|

|
| DC39103 | Remibrutinib (LOU064) Featured |
Remibrutinib (LOU064) is a potent, highly selective covalent inhibitor of bruton tyrosine kinase (BTK) with IC50 of 1.3 nM, 2.5 nM and 18 nM for BTK, FcγR-induced IL8 and anti-IgM/IL4-induced CD69, respectively. Remibrutinib (LOU064) exhibits an exquisite kinase selectivity due to binding to an inactive conformation of BTK and has the potential for the treatment of autoimmune diseases.
More description
|

|
| DC22619 | Rotigotine Featured |
A non-selective agonist of the dopamine D3 receptor (Ki=0.71 nM).
More description
|

|
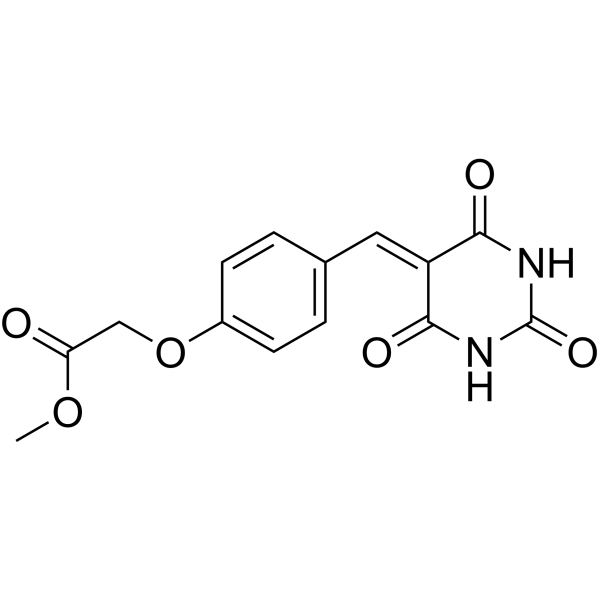
| DC65532 | MtUng-IN-1 Featured |
MtUng-IN-1 (Compound 18a) is a Uracil DNA glycosylase of Mycobacterium (MtUng) inhibitor (IC50: 300 μM). MtUng-IN-1 can be used for research of cancers and infectious diseases.
More description
|
|
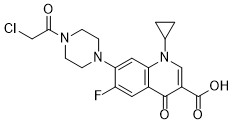
| DC65531 | Anticancer agent 118 Featured |
Anticancer agent 118, a N‑acylated ciprofloxacin derivative, has anti-bacterial and anticancer activities.
More description
|
|
| DC42414 | Zandelisib Featured |
Zandelisib is a phosphatidylinositol 3-kinase (PI3K) extracted from patent WO2019183226 A1, Compound Example 1. Zandelisib selectively inhibits p110δ with an IC50 of 3.5 nM. Zandelisib functions as an antineoplastic.
More description
|

|
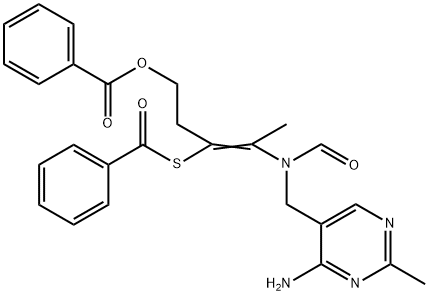
| DC36541 | Bentiamine Featured |
Bentiamine is also known as Dibenzoyl Thiamine. Dibenzoyl Thiamine (Bentiamine), a derivative of thiamine, is rapidly absorbed into the body and converted to thiamine.
More description
|
|
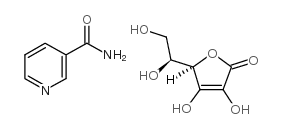
| DC65530 | niacinamide ascorbate Featured |
Nicotinamide ascorbate is a bioactive chemical.
More description
|
|
| DC70064 | Parabulin |
Parabulin is a novel potent, parasite-specific tubulin inhibitor, inhibits growth of parasites while displaying no effects on human cells.
More description
|
|
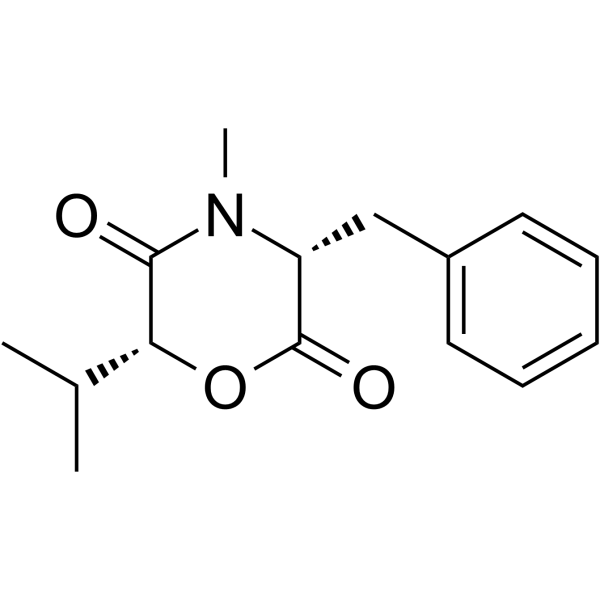
| DC60310 | (3S,6R)-Lateritin Featured |
Lateritin is a potent inhibitor of acyl-CoA:cholesterol acyltransferase (ACAT), isolated from the mycelial cake of Gibberella lateritium IFO 7188[1]. Lateritin also inhibits the growth of a mini-panel of human cancer cell lines, gram-positive bacteria, and Candida albicans[2].
More description
|
|
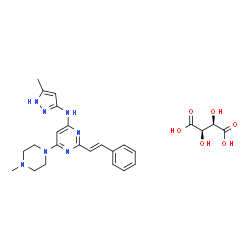
| DC50027 | ENMD-2076 L-(+)-Tartaric acid Featured |
ENMD-2076 Tartrate is a multi-targeted kinase inhibitor with IC50s of 1.86, 14, 58.2, 15.9, 92.7, 70.8, 56.4 nM for Aurora A, Flt3, KDR/VEGFR2, Flt4/VEGFR3, FGFR1, FGFR2, Src, PDGFRα, respectively.
More description
|
|
| DC28914 | AhR modulator-1 Featured |
AhR modulator-1 (compound 6-MCDF) is a selective and orally active aryl hydrocarbon receptor (AhR) modulator. AhR modulator-1 inhibits metastasis, in part, by inhibiting prostatic VEGF production prior to tumor formation. AhR modulator-1 also possess anti-estrogenic properties in rat uterus.
More description
|

|
| DC27035 | EIDD-2801(Molnupiravir) Featured |
Molnupiravir, also known as EIDD-2801 and MK-4482, is an orally bioavailable form of a highly potent ribonucleoside analog that inhibits the replication of multiple RNA viruses including SARS-CoV-2, the causative agent of COVID-19. EIDD-2801 has been show
More description
|

|
| DC23034 | Isochlorogenic acid C Featured |
4,5-Dicaffeoylquinic acid ( Isochlorogenic acid C) possesses potent hepatoprotective and anti-HBV effects.IC50 value:Target: Anti-hepatitis natural produce.In vitro: To study anti-hepatitis effect of isochlorogenic acid C, anti-apoptotic and anti-injury properties of test compound were evaluated. The results showed that test compound at concentrations of 10 to 100 μg/ml significantly reduced the caspase-3 and transformed growth factor β1 (TGFβ1) levels of the D-GalN-challenged hepatocytes. Also, test compound improved markedly cell viability of the D-GalN-injured hepatocytes and produced a maximum protection rate of 47.28% at a concentration of 100 μg/ml. Furthermore, test compound significantly inhibited productions of HBsAg and HBeAg. Its maximum inhibitory rates on the HBsAg and HBeAg expressions were 86.93 and 59.79%, respectively. In addition, test compound significantly induced the HO-1 expression of HepG2.2.15 cells [1]. In vivo:
More description
|

|
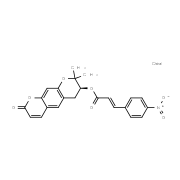
| DC21234 | LKY-047 |
LKY-047 is a decursin derivative that potently and selectively inhibits CYP2J2, competitively inhibits CYP2J2-mediated astemizole O-demethylase and terfenadine hydroxylase activity with Ki of 0.96 and 2.61 uM, respectively.
More description
|
|
| DC10887 | EPZ031686 Featured |
EPZ031686 is a noncompetitive inhibitor for SMYD3 and MEKK2 with a Ki=1.2 and 1.1 nM respectively.
More description
|
|
| DC72169 | RP-6685 Featured |
RP-6685 is a potent, selective and orally active DNA polymerase theta (Polθ) inhibitor with an IC50 value of 5.8 nM (PicoGreen assay). RP-6685 shows antitumor efficacy in mouse tumor xenograft model.
More description
|

|
| DC72808 | UCM-1306 Featured |
UCM1306 is a potent and orally active human dopamine D1 receptor allosteric modulator (PAM). UCM-1306 increases the endogenous dopamine (DA) maximal effect both in human and mouse D1 receptors. UCM-1306 is not only for improving motor symptoms but also for addressing the key comorbid cognitive impairment associated with long-term Parkinson’s disease (PD).
More description
|

|