 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
| Cat. No. | Product Name | Field of Application | Chemical Structure |
|---|---|---|---|
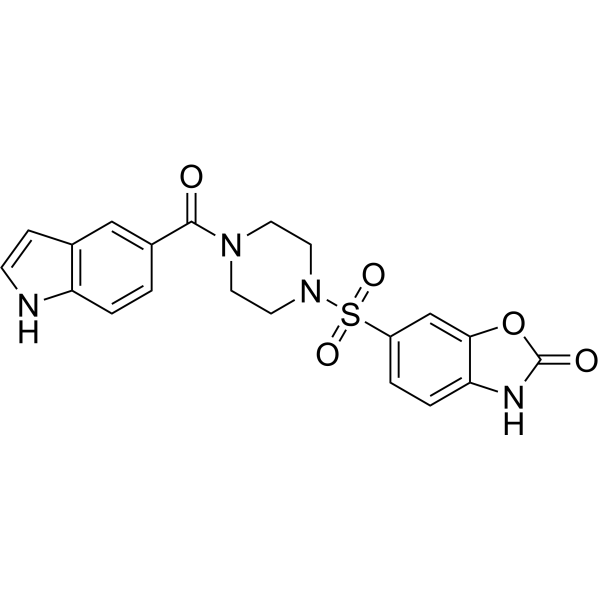
| DC72824 | TH1760 |
TH1760 is a first-in-class, potent, selective and cell-active NUDT15 (MTH2) inhibitor with an IC50 value of 25 nM.
More description
|
|
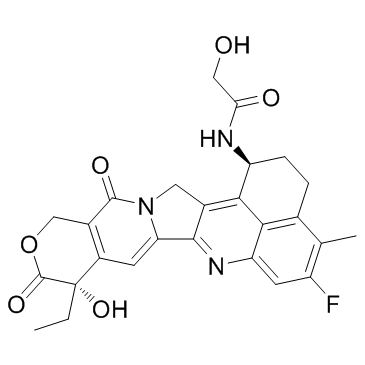
| DC65448 | Dxd Featured |
Dxd is a potent DNA topoisomerase I inhibitor, with an IC50 of 0.31 μM, used as a conjugated drug of HER2-targeting ADC (DS-8201a).
More description
|
|
| DC72861 | Diethyl Pyrocarbonate |
Diethyl pyrocarbonate is a potent, non-specific inhibitor of RNase. It has been useful as an in vitro agent, relatively specific for binding to imidazole of histidine. It inhibits central chemosensitivity in rabbit and can modify Ser, Thr, His and Tyr residues.
More description
|

|
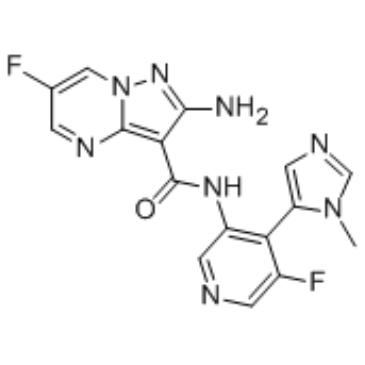
| DC65446 | ATR inhibitor 1 Featured |
ATR inhibitor 1 is a ATR inhibitor extracted from patent WO2015187451A1, compound I-l, has a Ki value below 1 µΜ[1].
More description
|
|
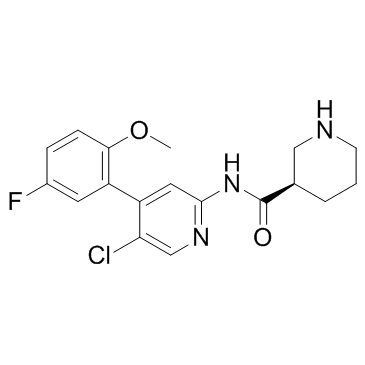
| DC9390 | CDK-IN-2 Featured |
CDK-IN-2 is a potent and sepecific CDK inhibitor.
More description
|
|
| DC70350 | Debio 0123 Featured |
Debio 0123 (Debio0123) is a potent, orally available, and highly selective Wee1 kinase inhibitor with IC50 of 0.8 nM, Ki of 0.1 nM.Debio 0123 is highly selective to Wee1 on 465 selected kinases in a cellfree system, does not inhibit Plk1 and Plk2, as also reported in recent publications for AZD1775.Debio 0123 exhibits in vitro growth inhibition activity in a broad number of human cancer cell lines (IC50= 0.109 to 7.08 uM).Debio 0123 prevent CDC2 / Cdk1 phosphorylation, induces γH2AX and enhances phosphorylation of histone H3.Debio 0123 induces apoptosis through mitotic catastrophe following cell cycle progression despite accumulation of unrepaired DNA damage both in vitro and in vivo.
More description
|

|
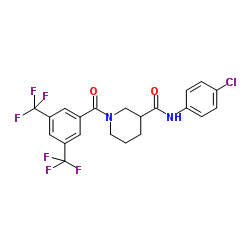
| DC45326 | CCG-100602 Featured |
CCG-100602 is a specific inhibitor of myocardin-related transcription factor A/serum response factor (MRTF-A/SRF) signaling. CCG-100602 specifically block MRTF-A nuclear localization and thus inhibit the fibrogenic transcription factor SRF.
More description
|
|
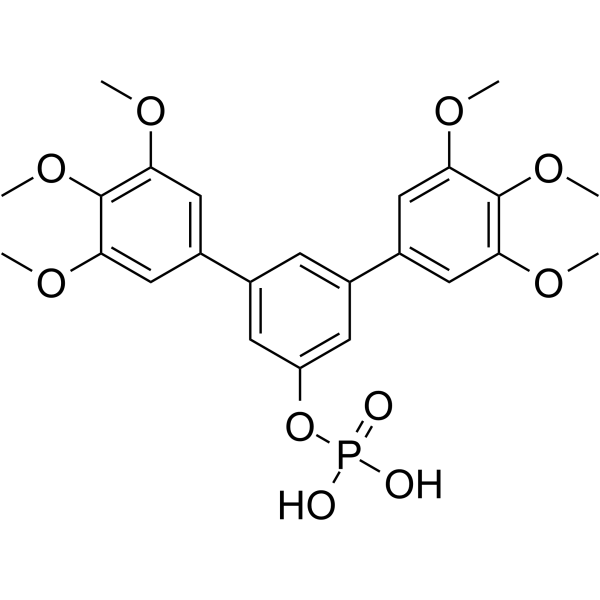
| DC65444 | Stafia-1 Featured |
Stafia-1 is the first STAT5a inhibitor that inhibits STAT5a (IC50=22.2 μM, Ki=10.9 μM) with at least 9‐fold selectivity over STAT5b and higher selectivity against other STAT family members.
More description
|
|
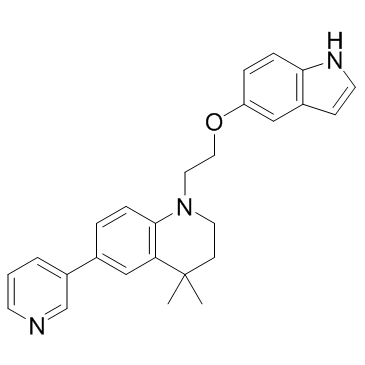
| DC21703 | STAT5 inhibitor 17f Featured |
A novel potent, selective inhibitor of phosphorylation and transcriptional activity of STAT5, but not STAT3, AKT, or Erk1/2 phosphorylation.
More description
|
|
| DC8726 | FLLL31 Featured |
FLLK31 is a potent and selective inhibitor of the STAT3 signaling pathway.
More description
|

|
| DC21702 | SC-99 Featured |
A novel selective STAT3 inhibitor that inhibits JAK2-STAT3 activation but has no effects on other transcription factors such as NF-κB, and kinases such as AKT, ERK, and c-Src.
More description
|

|
| DC11890 | Delgocitinib Featured |
Delgocitinib (JTE-052, LEO 124249) is a potent, selective, orally active, pan-JAK inhibitor with IC50 of 2.8, 2.6, 13 and 58 nM for JAK1,2,3 and Tyk2, respectively.
More description
|

|
| DC40041 | RSVA405 Featured |
RSVA405 is a potent, orally active activator of AMPK, with an EC50 of 1 μM. RSVA405 facilitates CaMKKβ-dependent activation of AMPK, inhibits mTOR, and promotes autophagy to increase Aβ degradation. RSVA405 has anti-inflammatory effects through the inhibition of STAT3 function. RSVA405 also can be used for the research of obesity.
More description
|

|
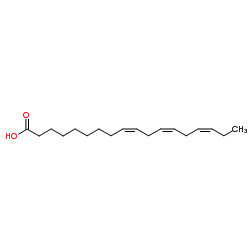
| DCY-109 | Linolenic acid Featured |
α-Linolenic acid, isolated from seed oils, is an essential fatty acid that cannot be synthesized by humans. α-Linolenic acid can affect the process of thrombotic through the modulation of PI3K/Akt signaling. α-Linolenic acid possess the anti-arrhythmic properties and is related to cardiovascular disease and cancer[1].
More description
|
|
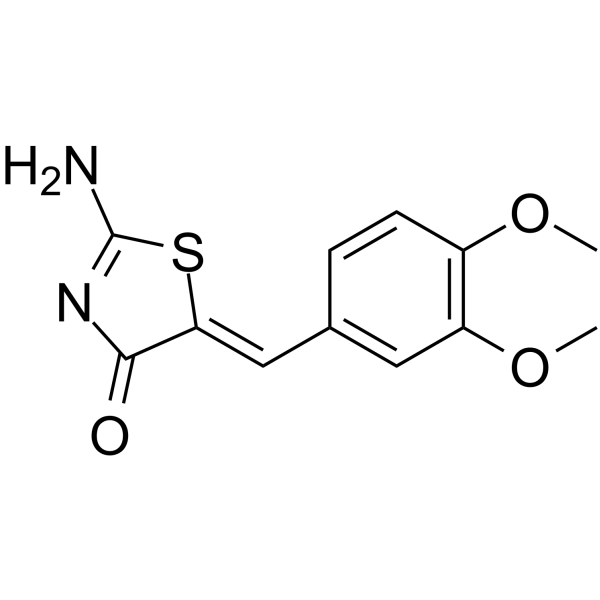
| DC65442 | WAY-119064 Featured |
way-119064 is a highly potent GSK-3β inhibitor with an IC50 value of 80.5 nM. GSK-3β inhibitor 10 can be used for researching Alzheimer’s disease[1].
More description
|
|
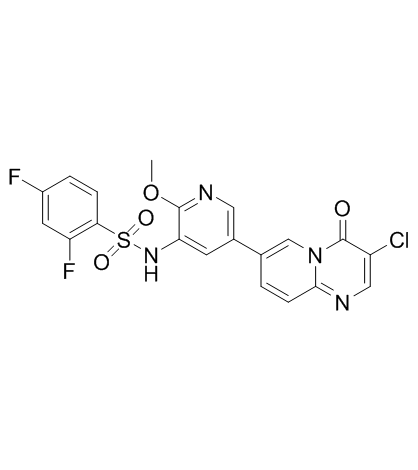
| DC20036 | PI3K/mTOR Inhibitor-2 Featured |
PI3K/mTOR Inhibitor-2 is a potent dual pan-PI3K/mTOR inhibitor with IC50s of 3.4/34/16/1 nM for PI3Kα/PI3Kβ/PI3Kδ/PI3Kγ and 4.7 nM for mTOR. Antitumor activity.
More description
|
|
| DC72738 | YLF-466D Featured |
YLF-466D is an AMPK activator that inhibits platelet aggregation with IC50 of 84 μM.
More description
|

|
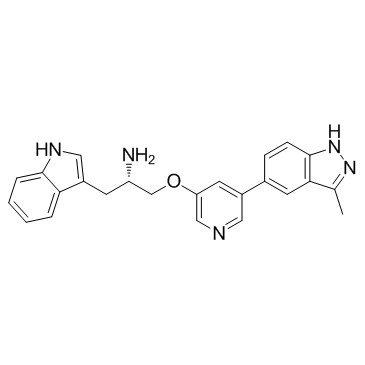
| DC22498 | A-443654 Featured |
A potent and selective Akt inhibitor with Ki of 0.16 nM for Akt1.
More description
|
|
| DC47381 | MOMIPP Featured |
MOMIPP, a macropinocytosis inducer, is a PIKfyve inhibitor. MOMIPP penetrates the blood-brain barrier (BBB).
More description
|

|
| DC65441 | Recilisib Featured |
Recilisib (ON01210, EX-RAD) is a radioprotectant that activates the activity of AKT and PI3K in cells. It has been studied as prophylactic (use prior to radiation exposure) and therapeutic (after exposure to radiation) drug.
More description
|

|
| DC1023 | CHIR-99021 (CT99021) Featured |
CHIR-99021 is a glycogen synthase kinase 3 beta inhibitor that has antiproliferative activity in vitro and in vivo.
More description
|

|
| DC57120 | PF07321332(nirmatrelvir) Featured |
PF07321332(nirmatrelvir) is a potent and orally active SARS-CoV 3C-like protease (3CLPRO) inhibitor . PF-07321332 targets to the SARS-CoV-2 virus and can be used for COVID-19 reseacrch.
More description
|

|
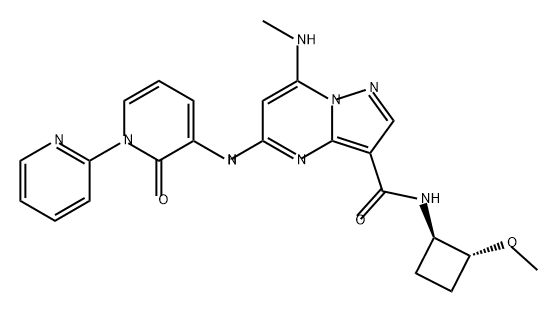
| DC72210 | NDI-034858(Zasocitinib) Featured |
NDI034858 is a TYK2 inhibitor, target TYK2 JH2 domain with binding constant Kd of <200 pM.
More description
|
|
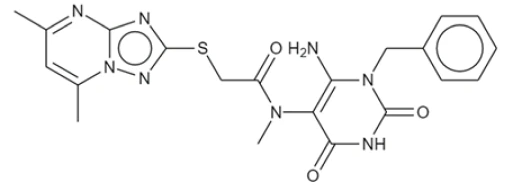
| DC60481 | SC9 Featured |
SC9 is an uncompetitive inhibitor of WT dynamin-related protein 1 (Drp1) with IC50 of 270 nM. SC9 completely prevents the LPS-induced decline in cells with fused mitochondria.
More description
|
|
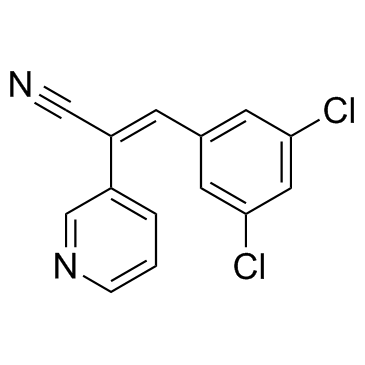
| DC10367 | RG14620 Featured |
RG14620 is an epidermal growth factor receptor (EGFR) inhibitor, with IC50 values of 3 μM for HER 14 colony formation and 1 pM for HER 14 DNA synthesis.
More description
|
|
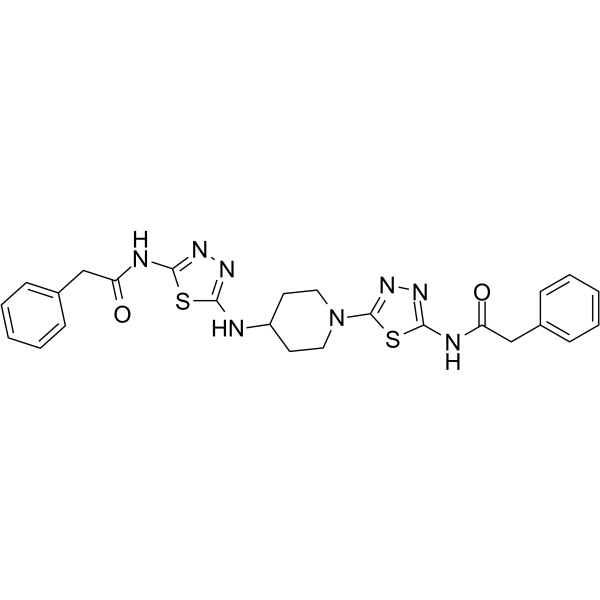
| DC20183 | UPGL00004 Featured |
UPGL00004 is a potent glutaminase C (GAC) inhibitor with an IC50 of 29 nM, showing high selectivity for GAC over GLS2.
More description
|
|
| DC10395 | S107 Featured |
S107 is a RyR-selective 1,4-benzothiazepine derivative that stabilizes RyR2 channels by enhancing the binding affinity of calstabin2 to mutant and/or PKA-phosphorylated channels.
More description
|
|
| DC7628 | PI-3065 Featured |
PI-3065 is a novel potent and selective PI3K p110δ inhibitor with IC50 of 15 nM; exhibits > 100 fold selectivity against p110α, p110β, p110γ, DNA-PK and mTOR.
More description
|

|
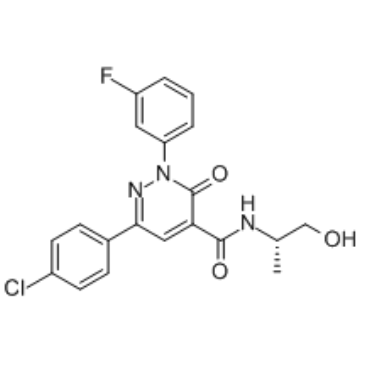
| DC26020 | BAY2335218(BAY-218) Featured |
BAY2335218(BAY-218) is the first-in-class AhR antagonist for overcoming tumor-mediated immunosuppression.
More description
|
|
| DC7761 | Brincidofovir (CMX-001) Featured |
CMX001 (Brincidofovir; HDP-CDV) was developed as an orally active, lipophilic form of cidofovir (CDV); has enhanced activity in vitro and in vivo compared to CDV against certain herpesviruses, adenoviruses and orthopoxviruses.IC50 Value: 5.5 nM (EC50, in PDA at 7 dpi) [3]Target: anti-CMVCMX001 is currently in Phase II clinical studies for development as a therapeutic agent for human CMV, adenovirus and BK virus infections, as well as, for adverse events following smallpox vaccinations.in vitro: In PDA at 7 dpi, the CMX001 50% effective concentration (EC50) was 5.55 nM, the 50% cytotoxic concentration (CC50) was 184.6 nM, and the 50% selectivity index (SI50) was 33.3. The EC90 was 19.7 nM, the CC90 was 5,054 nM, and the SI90 was 256.1. In COS-7 cells, JCV replication was faster and the EC50 and EC90 were 18- and 37-fold higher than those in PDA, i.e., 0.1 μM and 0.74 μM (CC50, 0.67 μM; SI50, 6.7; CC90, 12.2 μM; SI90, 16.5) at 5 dpi [3].in vivo: CMX001 and CDV are equally efficacious at protecting mice from mortality following high ectromelia virus doses (10,000 x LD(50)) introduced by the intra-nasal route or small particle aerosol. Using CMX001 at a 10mg/kg dose followed by 2.5mg/kg doses every other-day for 14 days provided solid protection against mortality and weight loss following an intra-nasal challenge of (100-200) x LD(50) of ectromelia virus [1]. When CMX001 was administered orally to mice infected with HSV-1, mortality was reduced significantly (p≤0.001) with all three dose levels when treatments were initiated 24 h post viral inoculation. When treatments were started 48 h post viral inoculation, 5 and 2.5 mg/kg significantly reduced mortality (p≤ 0.001). If treatments were delayed until 72 h post viral inoculation, CMX001 did not reduce mortality or increase the mean day to death. When mice were infected intranasally with HSV-1 and treatments initiated 24 h post viral inoculation using CMX001 at 5 mg/kg or ACV at 100 mg/kg, virus replication in target organs was reduced by both CMX001 and ACV when compared to vehicle treated mice [2]. Toxicity: Diarrhea was the most common adverse event in patients receiving CMX001 at doses of 200 mg weekly or higher and was dose-limiting at 200 mg twice weekly. Myelosuppression and nephrotoxicity were not observed [4].
More description
|

|