 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
| Cat. No. | Product Name | Field of Application | Chemical Structure |
|---|---|---|---|
| DC65467 | Kojic acid dipalmitate Featured |
Kojic acid dipalmitate (Kojic dipalmitate) is a derivative of Kojic acid (HY-W050154), a fungal metabolite that can be produced by species of Aspergillus, Acetobacter and Penicillium. Kojic acid dipalmitate is a slow and reversible competitive inhibitor of tyrosinase. Kojic acid dipalmitate can be used for skin‐lightening agent research[1].
More description
|

|
| DC65466 | Phenylthiourea Featured |
1-PHENYL-2-THIOUREA(N-Phenylthiourea, PTU, Phenylthiocarbamide) is an inhibitor of tyrosinase (Tyr). 1-PHENYL-2-THIOUREA blocks pigmentation and improves optical transparency in zebrafish (Danio rerio) embryo.
More description
|

|
| DC65465 | L-Ascorbic Acid Phospate Magnesium Salt Hydrate Featured |
L-ascorbic acid 2-phosphate (2-Phospho-L-ascorbic acid) magnesium hydrate is a long-acting vitamin C derivative that can stimulate collagen formation and expression. L-ascorbic acid 2-phosphate magnesium hydrate can be used as a culture medium supplement for the osteogenic differentiation of human adipose stem cells (hASCs). L-ascorbic acid 2-phosphate magnesium hydrate increases alkaline phosphatase (ALP) activity and expression of runx2A in hASCs during the osteogenic differentiation[1][2][3].
More description
|

|
| DC40756 | CBR-470-1 Featured |
CBR-470-1 is an inhibitor of the glycolytic enzyme phosphoglycerate kinase 1 (PGK1). CBR-470-1 is also a non-covalent Nrf2 activator. CBR-470-1 protects SH-SY5Y neuronal cells against MPP+-induced cytotoxicity through activation of the Keap1-Nrf2 cascade.
More description
|

|
| DC72892 | MEHP Featured |
MEHP (Phthalic acid mono-2-ethylexyl ester) is a competitive inhibitor of CYP2C9 with IC50 of 6.37 μM.
More description
|

|
| DC34053 | SM-7368 Featured |
SM-7368 inhibits TNF-alpha-induced MMP-9 upregulation in a concentration-dependent manner. SM-7368 strongly inhibits the TNF-alpha-induced invasion of HT1080 human fibrosarcoma cell line.
More description
|

|
| DC11926 | NDMC101 Featured |
A small molecule that inhibits NFATc1 and NF-κB activity, inhibits RANKL-induced osteoclastogenesis in vivo.
More description
|

|
| DC44511 | NF-κB-IN-1 Featured |
NF-κB-IN-1, a 4-arylidene crucumin analogue, is a potent NF-κB signaling pathway inhibitor. NF-κB-IN-1 directly inhibits IKK to block NF-κB activation. NF-κB-IN-1 effectively inhibits the viability of lung cancer cells and attenuates the clonogenic activity of A549 cells.
More description
|

|
| DC71677 | IMD-0560 Featured |
IMD-0560 is an Inhibitor of nuclear factor kappa-B kinase (IKK) which can suppress the production of inflammatory cytokines and chemokines.
More description
|

|
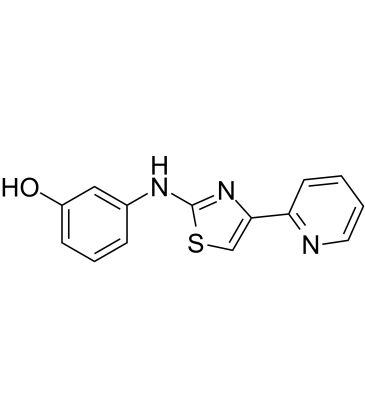
| DC72487 | KCC-07 Featured |
KCC-07 is a potent, selective and brain-penetrant inhibitor of MBD2 (methyl-CpG-binding domain protein 2) with anticancer activity. It prevents binding of MBD2 to methylated DNA and activates brain specific angiogenesis inhibitor 1 (BAI1) inducing anti-proliferative BAI1/p53/p21 signaling.
More description
|
|
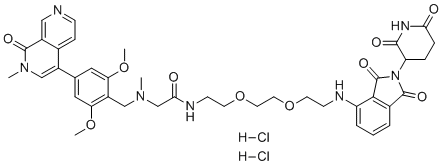
| DC70339 | dBRD9 dihydrochloride Featured |
dBRD9 dihydrochloride is a potent and selective degrader (PROTAC) of BRD9 with IC50 of 56.6 nM in MOLM-13 cells.dBRD9 is composed of the BRD9 inhibitor BI 7273 conjugated to the cereblon E3 ligase ligand pomalidomide.dBRD9 does not degrade BRD4 or BRD7 at concentrations up to 5 uM.dBRD9 exhibits antiproliferative effects in human AML cell lines.
More description
|
|
| DC72776 | SD49-7 Featured |
SD49-7 is an inhibitor of histone lysine-specific demethylase 4 (KDM4) with an IC50 of 0.19 µM.
More description
|

|
| DC72775 | iJMJD6 Featured |
iJMJD6(WL12) is an inhibitor of arginine demethylase JMJD6 with an IC50 value of 0.22 μM.
More description
|

|
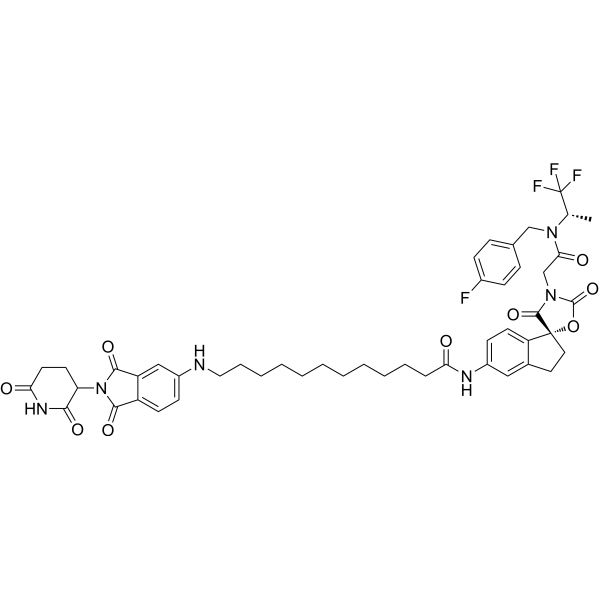
| DC72756 | JQAD1 Featured |
JQAD1 is a CRBN-dependent PROTAC that selectively targets EP300 for degradation. JQAD1 suppresses EP300 expression and the H3K27ac modification. JQAD1 induces apoptosis. JQAD1 can be used in research of cancer.
More description
|
|
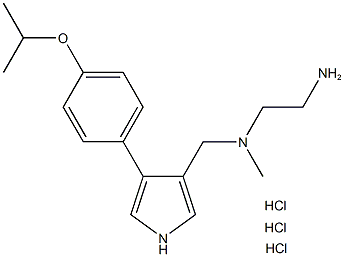
| DC72822 | MS023 hydrochloride Featured |
MS023 hydrochloride is a potent, selective and cell-active inhibitor of human type I protein arginine methyltransferases (PRMT), with IC50 values of 30, 119, 83, 4 and 5 nM for PRMT1, PRMT3, PRMT4, PRMT6, and PRMT8, respectively.
More description
|
|
| DC47467 | UZH2 Featured |
UZH2 is a potent and selective METTL3 inhibitor with an IC50 value of 5 nM.
More description
|

|
| DC22378 | DPH Featured |
A cell-permeable, small-molecule c-Abl kinase activator with pEC50 of 6.1(EC50=794 nM).
More description
|

|
| DC65459 | Enapotamab vedotin Featured |
Enapotamab vedotin (EnaV, HuMAX-AXL-ADC) is an AXL-specific human IgG1 antibody conjugated to the microtubule disrupting agent monomethyl auristatin E (MMAE) through a protease cleavable valine-citrulline (vc) linker.
More description
|

|
| DC65457 | Trastuzumab MMAE Featured |
Trastuzumab MMAE is an antibody-drug conjugate (ADC) composed of an anti-HER2 antibody, conjugated to Monomethyl Auristatin E (MMAE), via a Mc linker. Trastuzumab MMAE has the potential for use in research of solid tumors mainly in HER2-positive breast cancer.
More description
|
|
| DC65456 | Trastuzumab duocarmazine Featured |
Trastuzumab duocarmazine ((vic)-Trastuzumab duocarmazine) is a HER2-targeting ADC that is recognized and cleaved by histone B in tumor cells and selectively targets tumor cells. Trastuzumab duocarmazine has anti-tumor activity and can be used in cancer research related to uterine and ovarian sarcomas[1].
More description
|
|
| DC7786 | ELN 441958 Featured |
ELN-441958 is a potent, neutral antagonist of B1 receptor, inhibits the binding of the B1 agonist ligand [3H]DAKD to IMR-90 cells with Ki of 0.26 nM. ELN-441958 is highly selective for B1 over B2 receptors, and >500/ 2000-fold selective for the B1 over μ/δ-opioid receptor.IC50 value: 0.26 nM (Ki)Target: B1 Receptorin vitro: ELN-441958 is a novel small molecule bradykinin B1 receptor antagonist, based on the inhibition of agonist-induced increases in intracellular calcium in native and recombinant cells. ELN-441958 does not inhibit the activation of the human bradykinin B2 receptor at concentrations up to 10 μM, showing that it is highly selective for B1 over B2 receptors. ELN-441958 also displays good selectivity for B1 over other receptors examined in a broad screening panel. It is >500-fold and >2000-fold selective for the B1 receptor over the human μ- and δ-opioid receptor, the most potent off-target activity identified. In IMR-90 cells expressing the native human B1 receptor, ELN-441958 produced a concentration-dependent antagonism of the DAKD-induced calcium mobilization with a KB of 0.12 nM. [1]in vivo: ELN-441958 is essentially completely absorbed and produces high plasma levels after oral administration in rhesus monkeys.ELN-441958 has a moderate clearance and volume of distribution in both species following i.v. administration, consistent with the high metabolic stability in rat, rhesus, and human microsomes. ELN-441958 has high oral exposure and moderate plasma half-lives in rats and rhesus monkeys. The oral availability of ELN441958 in rats was 57%. ELN-441958 dose-dependently reduced carrageenan-induced thermal hyperalgesia in a rhesus monkey tail-withdrawal model, with an ED50 3 mg/kg s.c. [1]
More description
|
|
| DC9396 | Olcegepant Featured |
Olcegepant(BIBN 4096; BIBN 4096BS) is the first potent and selective non-peptide antagonist of the calcitonin gene-related peptide 1 (CGRP1) receptor(IC50= 0.03 nM).
More description
|

|
| DC65453 | Norepinephrine tartrate Featured |
Norepinephrine tartrate is a potent agonist of adrenergic receptor (AR). Norepinephrine activates α1, α2, β1 receptors.
More description
|
|
| DC65452 | Oleoyl-L-alpha-lysophosphatidic acid sodium salt Featured |
Oleoyl-L-alpha-lysophosphatidic acid sodium salt is an essential metabolite for membrane biosynthesis. LPA interacts with the G protein-coupled receptors (GPCRs), called the LPA receptor and mediates signaling. It acts as a endogenous agonist for LPA1 and LPA2 receptors.
More description
|
|
| DC12308 | VTX-27 Featured |
VTX-27 is a selective protein kinase C θ (PKC θ) inhibitor, with Kis of 0.08 nM and 16 nM for PKC θ and PKC δ.
More description
|
|
| DC32519 | SMAD3 Featured |
SMAD3 is a receptor-regulated intracellular protein that functions downstream of TGF-β and activin receptors and mediates their signaling, playing a role in cell proliferation, differentiation, apoptosis and formation of extracellular matrix. Smad3 Inhibitor, SIS3 is a cell-permeable pyrrolopyridine compound that selectively inhibits TGF-β1-dependent Smad3 phosphorylation and Smad3-mediated cellular signaling with no effect on Smad2, p38 MAPK, ERK, or PI 3-K signaling. It has been shown to reduce TGF-β1-induced type 1 procollagen expression and myofibroblast differentiation in normal dermal fibroblasts as well as scleroderma fibroblasts.
More description
|
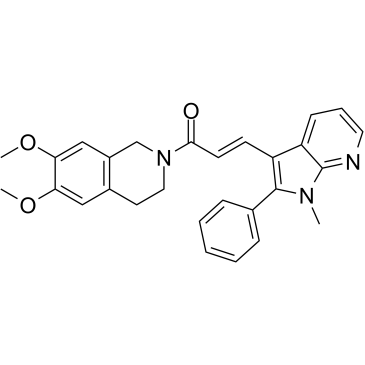
|
| DC28544 | N-Desmethyltamoxifen Featured |
N-Desmethyltamoxifen is the major metabolite of tamoxifen in humans. N-Desmethyltamoxifen, a poor antiestrogen, is a ten-fold more potent protein kinase C (PKC) inhibitor than Tamoxifen. N-Desmethyltamoxifen is also a potent regulator of ceramide metabolism in human AML cells, limiting ceramide glycosylation, hydrolysis, and sphingosine phosphorylation.
More description
|
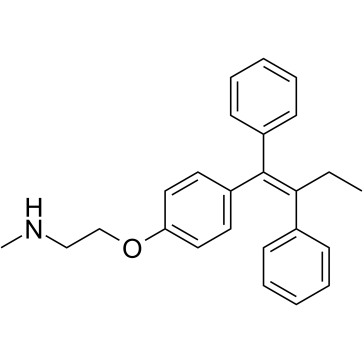
|
| DC65451 | Bisindolylmaleimide VIII (acetate) Featured |
Bisindolylmaleimide VIII acetate (Ro 31-7549 acetate) is a potent and selective protein kinase C (PKC) inhibitor with an IC50 of 158 nM for rat brain PKC. Bisindolylmaleimide VIII acetate has IC50s of 53, 195, 163, 213, and 175 nM for PKC-α, PKC-βI, PKC-βII, PKC-γ, PKC-ε, respectively[1]. Bisindolylmaleimide VIII acetate facilitates Fas-mediated apoptosis and inhibits T cell-mediated autoimmune diseases[2].
More description
|
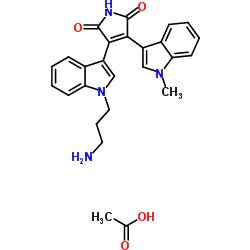
|
| DC65449 | ATM Inhibitor-5 Featured |
ATM Inhibitor-5 [formula (1)] is a potent inhibitor of serine/threonine protein kinase ATM (extracted from patent WO2022058351A1)[1].
More description
|
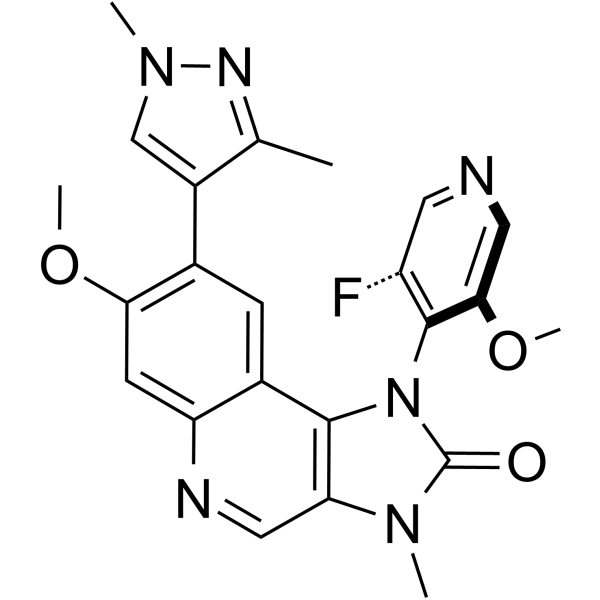
|
| DC72066 | 5-Ethynyluridine Featured |
5-Ethynyluridine (5-EU) is a potent cell-permeable nucleoside can be used to label newly synthesized RNA. 5-Ethynyluridine can be used for isolation and sequencing of nascent RNA from neuronal populations in vivo. 5-Ethynyluridine can be used to identify changes in transcription in vivo in nervous system disease models.
More description
|
|