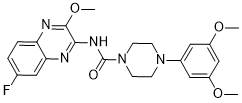
| In Vitro: |
RX-5902 (0-10 μM; 72 hours) is active against cell lines of all TNBC molecular subtypes and is active against cell lines with mutations in p53, RB1, CDKN2A, and loss of PTEN[1]. RX-5902 (20-100 nM; 24 hours) treatment results in a dose-dependent increase in tetraploid cells, consistent with induction of G2–M cell-cycle arrest[1]. RX-5902 (0-100 nM; 72 hours) exhibits no significant induction of apoptosis in cell lines resistant to the antiproliferative effects of RX5902. But in sensitive cells, the observed activation of apoptosis begins at 24–48 hours and reaches a peak at 72 hours. The induced apoptosis is greasted with a dose of 100 nM[1]. RX-5902 (0-100 nM; 24 or 48 hours) decreases MCL-1 expression as a dose-dependent manner in TNBC cell lines sensitive to RX-5902[1]. RX-5902 inhibits cell growth, MDA-MB-231, Caki-1, UMRC2, PANC-1, A549, MKN-45, HepG2, HCT116, HT29, PC-3, U251, HeLa, SK-MEL-28 and OVCAR-3 with IC50 values range from 0.01 μM to 0.021 μM in the growth inhibition of cancer cells[2]. Cell Viability Assay[1] Cell Line: MDA-MB-231, HCC1806, Hs578t, CAL-85-1, HCC38, HCC1187, MDA-MB-436, CAL-51, HCC38, BT549, MDAMB-157, HDQ-P1, HCC1395, MDA-MB-436, HCC1937, CAL-120, BT20 cells Concentration: 0-10 μM Incubation Time: 72 hours Result: Displayed the average IC50 of the cell lines sensitive to RX-5902 treatment is 56 nM. Cell Cycle Analysis[1] Cell Line: Sensitive (MDA-MB-231 and HCT1806) and two resistant (MDA-MB-436 and CAL-120) cell lines Concentration: 20 nM; 100 nM Incubation Time: 24 hours Result: Led to G2-M cell-cycle arrest at sensitive cells. Apoptosis Analysis[1] Cell Line: Sensitive (MDA-MB-231 and HCT1806) and two resistant (MDA-MB-436 and CAL-120) cell lines Concentration: 0-100 nM Incubation Time: 24-72 hours Result: Induced cell apoptosis in sensitive cell lines and peaks at 72 hours. Western Blot Analysis[1] Cell Line: Cal-51, HCC-1806, and MDA-MB-468 cells Concentration: 20 nM; 100 nM Incubation Time: 24 hours Result: Induced inhibition of MCL-1 expression in Cal-51, HCC-1806, and MDA-MB-468 cells. |
| References: |
[1]. Kost GC, et al. A Novel Anti-Cancer Agent, 1-(3,5-Dimethoxyphenyl)-4-[(6-Fluoro-2-Methoxyquinoxalin-3-yl)Aminocarbonyl] Piperazine (RX-5902), Interferes With β-Catenin Function Through Y593 Phospho-p68 RNA Helicase.J Cell Biochem. 2015 Aug;116(8):1595-601.
[2]. Capasso A, et al. First-in-Class Phosphorylated-p68 Inhibitor RX-5902 Inhibits β-Catenin Signaling and Demonstrates Antitumor Activity in Triple-Negative Breast Cancer.Nov;18(11):1916-1925. |
 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.