 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
| Cat. No. | Product Name | Field of Application | Chemical Structure |
|---|---|---|---|
| DC20247 | (R,R)-BNC375 Featured |
(R,R)-BNC375 is a potent, selective, and orally available type I positive allosteric modulator of α7 nAChRs.
More description
|

|
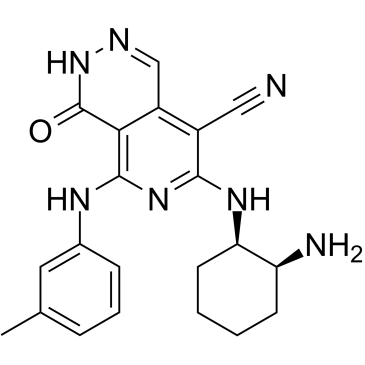
| DC65280 | DS21360717 Featured |
DS21360717 is a potent and orally active FER tyrosine kinase inhibitor, with an IC50 of 0.49 nM. Anti-cancer activity.
More description
|
|
| DC65279 | NPD2381 Featured |
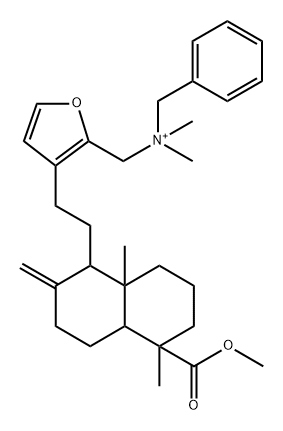
|
|
| DC70655 | NLX-204 Featured |
NLX-204 is a potent and selective ERK1/2 phosphorylation-preferring serotonin 5 HT1A receptor agonist with pKi = 10.19. NLX-204 displayed high selectivity in the SafetyScreen44 panel (including hERG channel), high solubility, metabolic stability, and Caco-2 penetration and did not block CYP3A4, CYP2D6 isoenzymes, or P-glycoprotein. Preliminary in vivo studies confirmed its promising pharmacokinetic profile. NLX-204 also robustly stimulated ERK1/2 phosphorylation in rat cortex and showed highly potent (MED = 0.16 mg/kg) and efficacious antidepressant-like activity, totally eliminating immobility in the rat Porsolt test.
More description
|
.png)
|
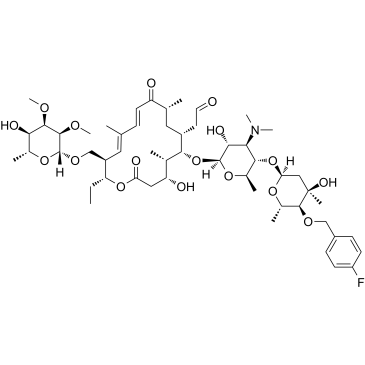
| DC65278 | ABBV-4083(Compound5d) Featured |
ABBV-4083 is an analog of Tylosin A that has potent anti-Wolbachia and anti-filarial activity.
More description
|
|
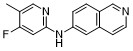
| DC65277 | JNJ-64326067 Featured |
JNJ-64326067 is a potent and selective binder to aggregated tau with a favorable pharmacokinetic profile and no apparent off-target binding. This was confirmed in rat and monkey positron emission tomography studies using [18F]-JNJ-64326067.
More description
|
|
| DC23532 | BMS-986166 Featured |
BMS-986166 (BMS986166) is a potent, selective S1P receptor modulator for the treatment of ulcerative colitis..
More description
|

|
| DC21989 | AES-135 Featured |
AES-135 (AES135) is a novel HDAC inhibitor that biochemically inhibits HDACs 3, 6, 8 and 11 with IC50 values of 190-1100 nM.
More description
|

|
| DC46443 | EML741 Featured |
EML741 is a histone lysine methyltransferase G9a/GLP inhibitor, with an IC50 of 23 nM, Kd of 1.13 μM for G9a. EML741 also inhibits DNMT1 (IC50, 3.1 μM), with no effect on DNMT3a or DNMT3b. EML741 exhibits low cell toxicity, and is membrane permeable and blood-brain barrier penetrated.
More description
|
.png)
|
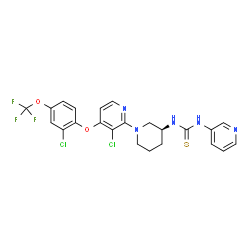
| DC65276 | (S)-DO271 Featured |
(S)-DO271 is an inactive control for the α/β-hydrolase domain-containing protein 12 (ABHD12) inhibitor DO264.
More description
|
|
| DC23570 | MIDD0301 Featured |
MIDD0301 is a potent, positive allosteric, α5β3γ2 selective, GABAA receptor (GABAAR) ligand with EC50 of 17 nM, shows no significant binding at the peripheral GABAAR at 10 uM.
More description
|

|
| DC20052 | CB-6644 Featured |
CB-6644 is a selective inhibitor of RUVBL1/2 complex with anti-cancer activity. CB-6644 blocks the ATPase activity of RUVBL1/2 with an IC50 of 15 nM.
More description
|

|
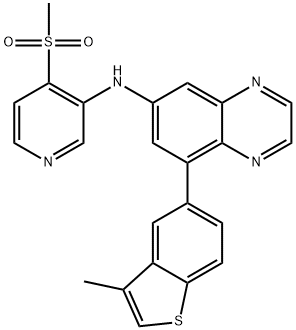
| DC65275 | PFKFB3inhibitor(Compound69) Featured |
|
|
| DC72617 | LAS190792 Featured |
LAS190792 (AZD8999) is a potent muscarinic antagonist and β2-adrenoceptor agonist with pIC50 8.9, 8.8, 8.8, 9.2, 8.2, 7.5, 9.1, 5.6 for M1, M2, M3, M4, M5, β1, β2, β3, respectively. LAS190792 can be used as a bronchodilator.
More description
|
.png)
|
| DC11551 | Elenbecestat Featured |
Elenbecestat (E2609) is a novel potent, oral BACE1 inhibitor for treatment of Alzheimer's disease..
More description
|

|
| DC11105 | Umibecestat Featured |
beta-secretase inhibitor.
More description
|

|
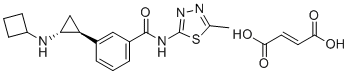
| DC12606 | T-448 Featured |
T-448 (T448) is a specific inhibitor of LSD1 enzyme activity, enhances H3K4 methylation in primary cultured rat neurons but has little impact on LSD1-GFI1B complex in human TF-1a erythroblasts.
More description
|
|
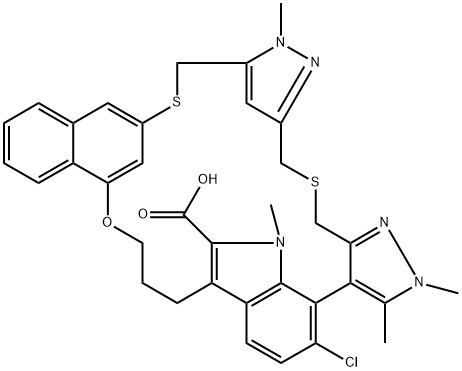
| DC20729 | AZD 5991 Featured |
AZD 5991 (AZD5991) is a potent and selective macrocyclic inhibitor of Mcl-1 with sub-nanomolar affinity.
More description
|
|
| DC11174 | ASP7657 Featured |
ASP7657 (ASP-7657) is a potent, selective, orally active prostaglandin EP4 receptor antagonist with Ki values of 6.02 nM and 2.21 nM for rat and human EP4 receptors, resepctively.
More description
|

|
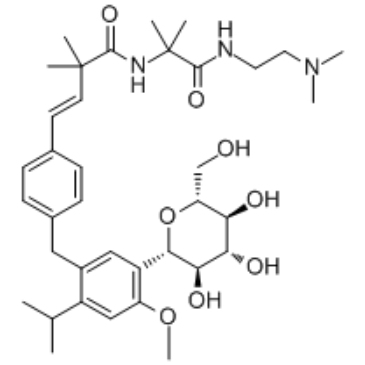
| DC65274 | SGL5213 Featured |
SGL5213 is a potent, oral active and low-absorbable sodium-dependent glucose cotransporter 1 (SGLT1) inhibitor, with IC50 values of 29 nM and 20 nM for hSGLT1 and hSGLT2, respectively. SGL5213 has potential to treat type 2 diabetes treatment.
More description
|
|
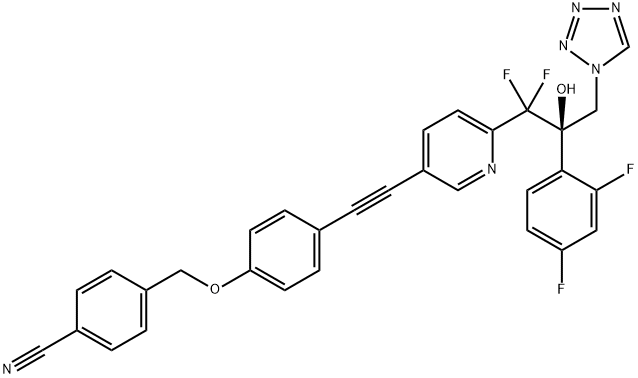
| DC23248 | VT-1598 Featured |
VT-1598 is a potent, high-affinity, oral inhibitor of fungal sterol 14α-demethylase (CYP51B) with Kd of 13 nM.
More description
|
|
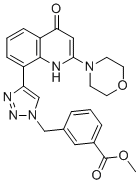
| DC12595 | CL27c Featured |
CL27c is a cell-permeable, inhaled lipophilic ester prodrug of CL27e, which is a potent, selective pan-class I PI3K inhibitor, CL27c is inactive in PI3K enzymatic assay, once inside the cytoplasm, CL27c is metabolized by unspecific esterases into CL27e.
More description
|
|
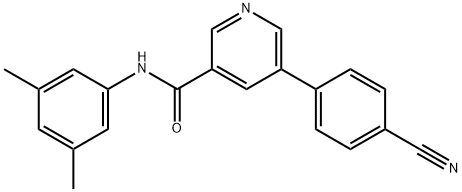
| DC65273 | Compound 16 Featured |
|
|
| DC20649 | AKB-9778 Featured |
AKB-9778 is a potent, selective inhibitor of vascular endothelial protein tyrosine phosphatase (VE-PTP) with IC50 of 17 pM.
More description
|
|
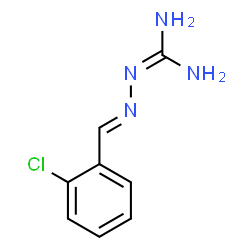
| DC8600 | Sephin-1 Featured |
Sephin1 is a selective inhibitor of stress-induced PPP1R15A and targets disease associated with accumulation of misfolded protein.
More description
|
|
| DC70739 | RMC-4630 Featured |
RMC-4630 is a potent and selective inhibitor of SHP2, a central node in the RAS signaling pathway.
More description
|

|
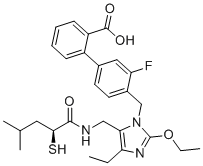
| DC12587 | TD-0212 Featured |
TD-0212 (TD0212) is a potent, orally efficacious, dual AT1 antagonist/Neprilysin (NEP) inhibitor with pKi of 8.9 (AT1) and pIC50 of 9.2 (NEP).
More description
|
|
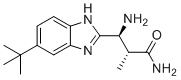
| DC12594 | PF-06305591 Featured |
PF-06305591 (PF06305591) is a potent, highly selective selective NaV1.8 blocker with IC50 of 15 nM, displays no significant activity against other sodium channel subtypes, K+ channels and Ca2+ channels.
More description
|
|
| DC8404 | E3330 Featured |
E3330 is a potent and selective APE1(Ref-1) inhibitor, which suppressed NF-kappa B DNA-binding activity.
More description
|

|
| DC11770 | PC786 Featured |
PC786 (PC-786) is a potent non-nucleoside RSV L-protein polymerase inhibitor with IC50 of 2.1 nM and 0.5 nM in cell-free enzyme assay and mini-genome assay in HEp-2 cells, respectively.
More description
|

|