 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
| Cat. No. | Product Name | Field of Application | Chemical Structure |
|---|---|---|---|
| DC8078 | PD 153035(AG-1517) Featured |
PD 153035(AG-1517) is an ultra-potent inhibitor of epidermal growth factor receptor tyrosine kinase (EGFRK), with an IC50 of 25 pM.
More description
|
|
| DC8283 | PD 150606 Featured |
PD 150606 is a selective, cell-permeable non-peptide calpain inhibitor (Ki values for ν and m-calpains are 0.21 and 0.37 μM respectively).
More description
|
|
| DC1106 | PCI-32765 (Ibrutinib) Featured |
PCI-32765 (Ibrutinib) is a potent and highly selective Btk inhibitor with IC50 of 0.5 nM.
More description
|

|
| DC10757 | PCI-29732 Featured |
PCI 29732 is a selective and irreversible Btk inhibitor with IC50 of 8.2 nM in a FRET based biochemical enzymology assay.
More description
|

|
| DC9723 | PBTZ169 Featured |
PBTZ169 is a decaprenyl-phosphoribose-epimerase (DprE1) inhibitor.
More description
|

|
| DC7550 | PBIT Featured |
PBIT is a potent, cell-permeable inhibitor of Jumonji histone demethylase (JHDM). Innhibits JARID1B (also known as KDM5B or PLU1) with an IC₅₀ of about 3 μm in vitro.
More description
|
|
| DC9776 | Pazopanib Hydrochloride Featured |
Pazopanib Hcl (GW-786034) is a novel multi-target inhibitor of VEGFR1, VEGFR2, VEGFR3, PDGFR, FGFR, c-Kit and c-Fms with IC50 of 10 nM, 30 nM, 47 nM, 84 nM, 74 nM, 140 nM and 146 nM, respectively.
More description
|

|
| DC11445 | Parthenolide Featured |
Parthenolide is a sesquiterpene lactone from the plant feverfew (T. parthenium).
More description
|
|
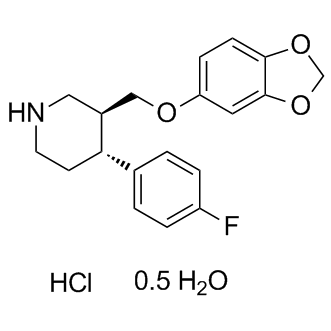
| DC9098 | Paroxetine HCl Featured |
Paroxetine hydrochloride hemihydrate is a antidepressant agents known as selective serotonin-reuptake inhibitors (SSRIs).
More description
|
|
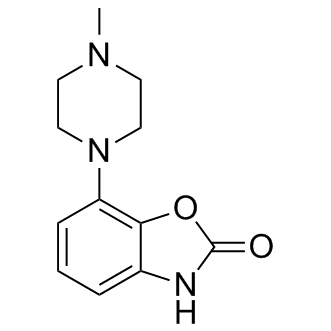
| DC6911 | pardoprunox (SLV308) Featured |
Pardoprunox(SLV-308) is a novel partial dopamine D2 and D3 receptor agonist and serotonin 5-HT1A receptor agonist; D2 (pKi = 8.1) and D3 receptor (pKi = 8.6) partial agonist (IA = 50% and 67%, respectively) and 5-HT1A receptor (pKi = 8.5) full agonist (IA
More description
|
|
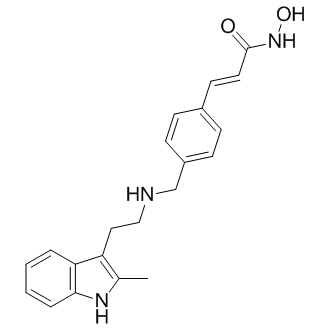
| DC7183 | Panobinostat(LBH589) Featured |
Panobinostat(LBH-589) is a broad-spectrum HDAC inhibitor; low nanomolar concentrations (IC50=5-20 nM) of LBH589 induced cell-cycle arrest, apoptosis, and histone (H3K9 and H4K8) hyperacetylation in MOLT-4 and Reh cell.
More description
|
|
| DC8459 | Palovarotene(R 667) Featured |
Palovarotene (R-667, RO-3300074) is a selective retinoic acid receptor gamma(RAR-γ) agonist for the treatment of emphysema.
More description
|

|
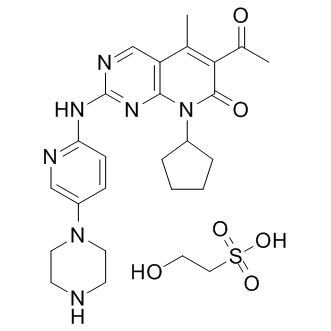
| DC8470 | Palbociclib isethionate Featured |
Palbociclib isethionate(PD-0332991 isethionate) is a highly specific inhibitor of Cdk4 (IC50=11 nM) and Cdk6 (IC50=16 nM), having no activity against a panel of 36 additional protein kinases.
More description
|
|
| DC5067 | Palbociclib (PD0332991 HCl) Featured |
Palbociclib is an oral, small molecule cyclin-dependent kinase 4/6 (CDK4/6) inhibitor, now in Phase 3 clinical development for advanced breast cancer.
More description
|

|
| DC8469 | Palbociclib Featured |
Palbociclib (PD-0332991) is an orally available pyridopyrimidine-derived cyclin-dependent kinase (CDK) inhibitor with potential antineoplastic activity.
More description
|

|
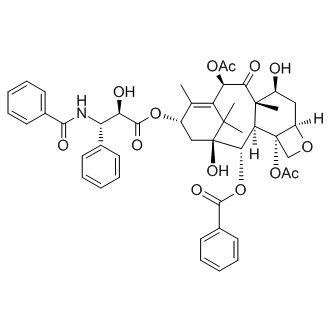
| DC8638 | Paclitaxel Featured |
Paclitaxel is a microtubule polymer stabilizer with IC50 of 0.1 pM in human endothelial cells.
More description
|
|
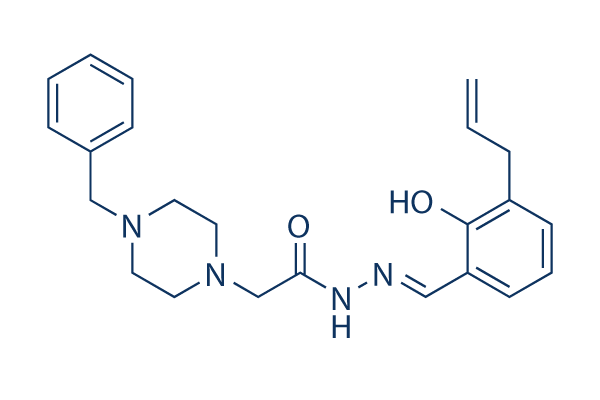
| DC7477 | pac-1 Featured |
PAC-1 is a potent procaspase-3 activator with EC50 of 0.22 μM and the first small molecule known to directly activate procaspase-3 to caspase-3.
More description
|
|
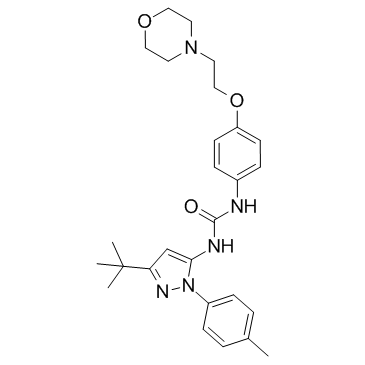
| DC10798 | p38-α MAPK-IN-1 Featured |
p38-α MAPK-IN-1 is an inhibitor of MAPK14 (p38-α), with IC50 of 2300 nM in EFC displacement assay, and 5500 nM in HTRF assay.
More description
|
|
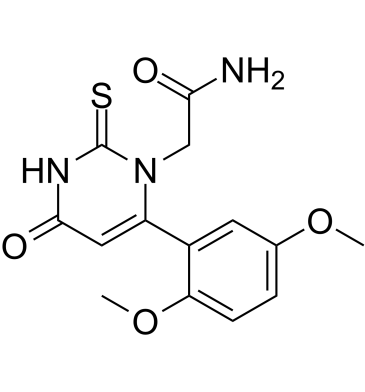
| DC25200 | PF-1355 Featured |
PF-1355 is a potent, selective, orally active Myeloperoxidase (MPO) inhibitor with EC50 of 1.47 uM.
More description
|
|
| DC12012 | PD 404182 Featured |
PD 404182 is a high affinity inhibitor of KDO 8-P synthase (Ki = 26 nM). Also inhibits dimethylarginine dimethylaminohydrolase 1 (DDAH1). Exhibits antiangiogenic and antiviral activity in vitro. Putative antibiotic against gram-negative bacteria.
More description
|

|
| DC47315 | ML-SI3 Featured |
ML-SI3 is a potent TRPML channel inhibitor with IC50s of 4.7 µM and 1.7 µM for TRPML1 and TRPML2, respectively.
More description
|

|
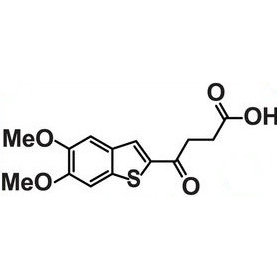
| DC39031 | MSA-2 Featured |
MSA-2 is an orally available human STING agonist.MSA-2 is bound to STING as a noncovalent dimer. Extensive experimental analysis indicates that MSA-2 predimerization is required for binding. Acidic tumor microenvironments favor permeable, uncharged MSA-2.
More description
|
|
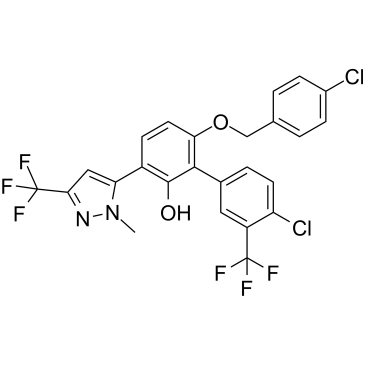
| DC32580 | MYCi975 Featured |
MYCi975, also known as NUCC-0200975, is a potent and selective MYC Inhibitor. MYCi975 disrupts MYC/MAX interaction, promotes MYC T58 phosphorylation and MYC degradation, and impairs MYC driven gene expression.
More description
|
|
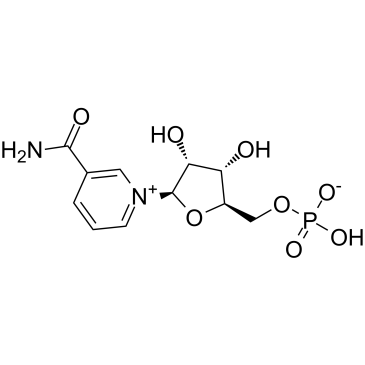
| DC32374 | Nicotinamide Mononucleotide Featured |
Nicotinamide Mononucleotide, also known as ("NMN" and "β-NMN") is a nucleotide derived from ribose and nicotinamide Like nicotinamide riboside, NMN is a derivative of niacin, and humans have enzymes that can use NMN to generate nicotinamide adenine dinuc
More description
|
|
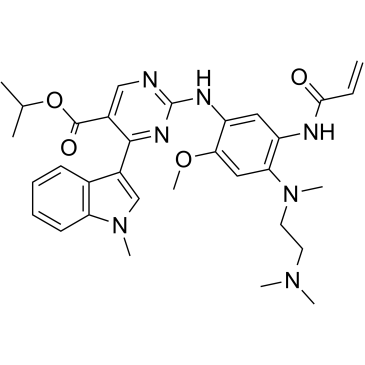
| DC28953 | Mobocertinib(TAK-788) Featured |
Mobocertinib is a epidermal growth factor receptor (EGFR) inhibitor and an antineoplastic.
More description
|
|
| DC28545 | N-Desmethyltamoxifen hydrochloride Featured |
N-Desmethyltamoxifen hydrochloride is the major metabolite of tamoxifen in humans. N-Desmethyltamoxifen, a poor antiestrogen, is a ten-fold more potent protein kinase C (PKC) inhibitor than Tamoxifen. N-Desmethyltamoxifen hydrochloride is also a potent re
More description
|

|
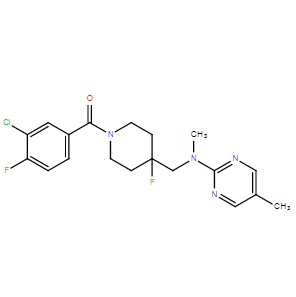
| DC26175 | NLX-101(F-15599) Featured |
NLX-101(F-15599) is a novel compound that activates serotonin 5-HT1A receptors with exceptional selectivity, having over 1000-fold higher affinity for this target over other receptors.
More description
|
|
| DC11363 | NSC 185058 Featured |
NSC 185058 is an inhibitor of ATG4B, a cysteine protease that activates LC3B during the initiation of autophagy.
More description
|

|
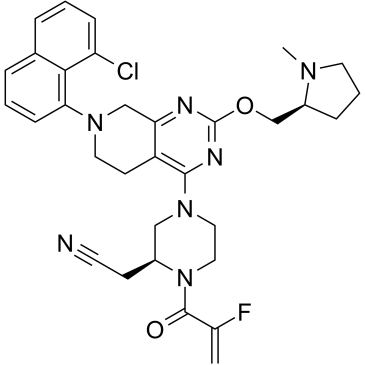
| DC26136 | MRTX849(Adagrasib) Featured |
MRTX849(Adagrasib) is a potent, orally-available, and mutation-selective covalent inhibitor of KRAS G12C with potential antineoplastic activity. MRTX849 covalently binds to KRAS G12C at the cysteine at residue 12, locks the protein in its inactive GDP-bou
More description
|
|
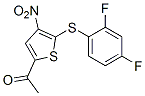
| DC7221 | P22077 Featured |
P22077 is a potent inhibitor of ubiquitin-specific protease (USP) 7 (EC50=8.6 uM), P22077 also inhibits the closely related deubiquitinase (DUB) USP47.
More description
|
|