 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
| Cat. No. | Product Name | Field of Application | Chemical Structure |
|---|---|---|---|
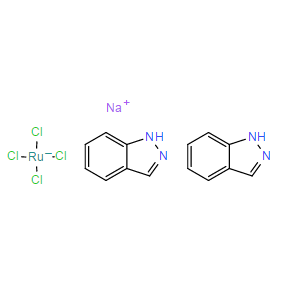
| DC9786 | NKP-1339 Featured |
NKP-1339 is the first-in-class ruthenium-based anticancer drug in clinical development against solid cancer and has recently been studied successfully in a phase I clinical trial.
More description
|
|
| DC9561 | Nitisinone Featured |
Nitisinone(SC0735) is an inhibitor of the enzyme 4-hydroxyphenylpyruvate dioxygenase.
More description
|

|
| DC8608 | BIBF 1120 esylate Featured |
Nintedanib (BIBF 1120) is a potent triple angiokinase inhibitor for VEGFR1/2/3, FGFR1/2/3 and PDGFRα/β with IC50 of 34 nM/13 nM/13 nM, 69 nM/37 nM/108 nM and 59 nM/65 nM.
More description
|

|
| DC7084 | Nintedanib (BIBF 1120) Featured |
Nintedanib (BIBF 1120) is a potent triple angiokinase inhibitor for VEGFR1/2/3, FGFR1/2/3 and PDGFRα/β with IC50 of 34 nM/13 nM/13 nM, 69 nM/37 nM/108 nM and 59 nM/65 nM.
More description
|

|
| DC3144 | Nilotinib Featured |
Nilotinib (AMN-107, Tasigna) is a Bcr-Abl inhibitor with IC50 less than 30 nM.
More description
|

|
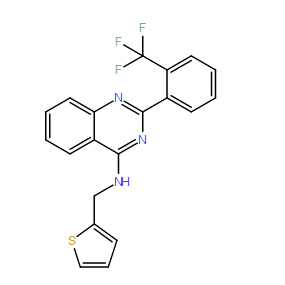
| DC10028 | NIH-12848 Featured |
NIH-12848 (NCGC00012848-02) is a putative phosphatidylinositol 5-phosphate 4-kinase γ (PI5P4Kγ) inhibitor, was explored as a tool for investigating this enigmatic, low activity,
More description
|
|
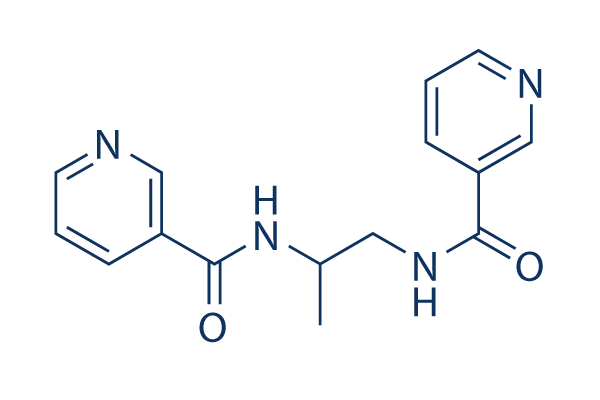
| DC7977 | Nicaraven Featured |
Nicaraven is an antivasospastic substance.
More description
|
|
| DC8836 | NIBR189 Featured |
NIBR 189 is a potent and selective EBI2 (GPR183) receptor antagonist (IC50 values are 11 and 15 nM for human and mouse EBI2 receptors respectively).
More description
|

|
| DC8076 | NG 52 (Compound 52 ) Featured |
NG 52 (Compound 52) is a potent, cell-permeable, reversible, selective, and ATP-compatible inhibitor of the cell cycle-regulating kinase, Cdc28p (IC50 = 7 μM), and the related Pho85p kinase (IC50 = 2 μM).
More description
|

|
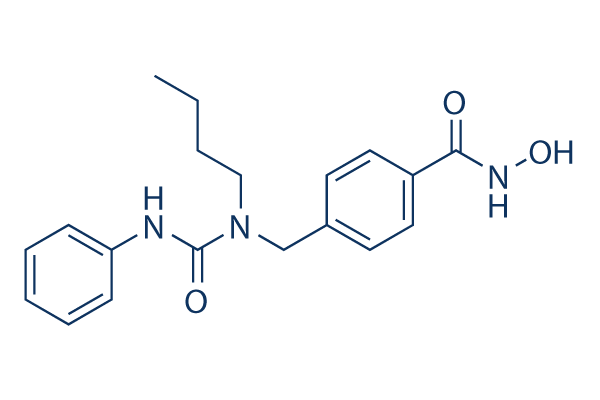
| DC7548 | Nexturastat A Featured |
Nexturastat A is a potent and selective HDAC6 inhibitor with IC50 of 5 nM, >190-fold selectivity over other HDACs.
More description
|
|
| DC9019 | Nevirapine |
Nevirapine is a potent, non-nucleoside reverse transcriptase inhibitor (NNRTI) used in combination with nucleoside analogues for treatment of HIV-1 infection and AIDS.
More description
|

|
| DC7211 | Neratinib (HKI-272) Featured |
Neratinib (HKI-272) is an orally available, irreversible tyrosine kinase inhibitor with IC50 of 59 nM and 92 nM for HER2 and EGFR, respectively.
More description
|

|
| DCAPI1393 | Nedaplatin (Aqupla) Featured |
Nedaplatin (Aqupla) is a derivative of cisplatin for inhibition of tumor colony forming units with IC50 of 28.5 μg/mL.
More description
|

|
| DC9817 | Necrosulfonamide (NSA) Featured |
Necrosulfonamide (NSA) is a very specific and potent necrosis inhibitor with an IC50 less than 0.2 uM.
More description
|

|
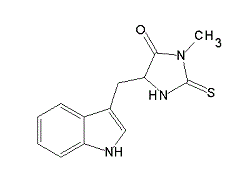
| DC2084 | Necrostatin-1 Featured |
Necrostatin-1 is a specific RIP1 inhibitor and inhibits TNF-α-induced necroptosis with EC50 of 490 nM.
More description
|
|
| DC10455 | NE-100 Featured |
NE100 hydrochloride is a potent and selective σ1 receptor antagonist (Ki = 0.86 nM) that displays > 55-fold selectivity over σ2 receptors and > 6000-fold selectivity over D1,
More description
|

|
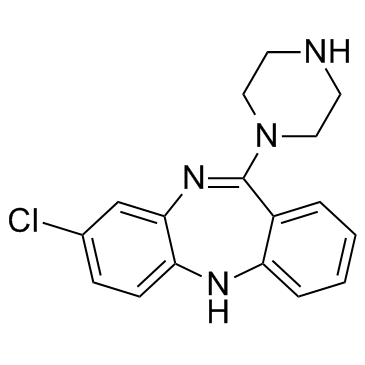
| DC10438 | N-Desmethylclozapine Featured |
N-Desmethylclozapine is a dengue virus inhibitor, and an agonist of δ-opioid receptor.
More description
|
|
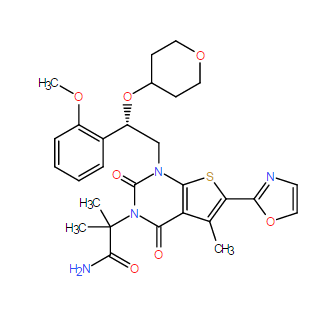
| DC9810 | ND-646 Featured |
ND-646(ND646) is a small-molecule allosteric inhibitor of acetyl-CoA carboxylase (ACC).
More description
|
|
| DC10173 | Firsocostat(ND-630,GS-0976) Featured |
ND-630 is an acetyl-CoA carboxylase (ACC) inhibitor; inhibits human ACC1 and ACC2 with IC50 values of 2.1 and 6.1 nM, respectively.
More description
|

|
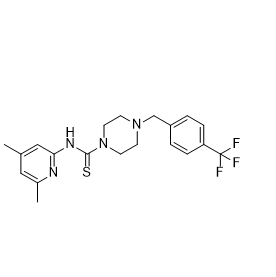
| DC10486 | NCT-503 Featured |
NCT-503 is an inhibitor of 3-phosphoglycerate dehydrogenase (PHGDH), inhibiting serine synthesis from 3-phosphoglycerate in cells with an IC50 value of 2.5 µM.
More description
|
|
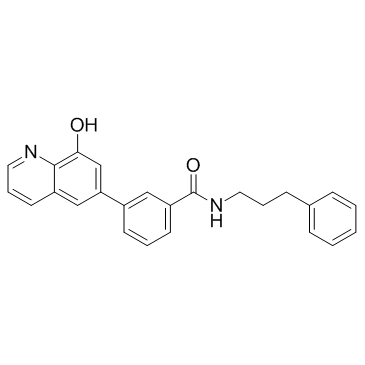
| DC10476 | NCGC00244536 Featured |
NCGC00244536 is a potent KDM4A inhibitor with an IC50 of 10 nM.
More description
|
|
| DC9770 | NCB-0846 Featured |
NCB-0846 is a novel,first-in-class,orally TNIK inhibitor with an IC50 value of 21 nM,that have shown strong anti-tumor efficacy against several cancer models.
More description
|

|
| DC8474 | Napabucasin (BBI608) Featured |
Napabucasin (BBI608) is an orally-administered small molecule which can block cancer stem cell (CSC) self-renewal and induces cell death in CSCs as well as non-stem cancer cells.
More description
|

|
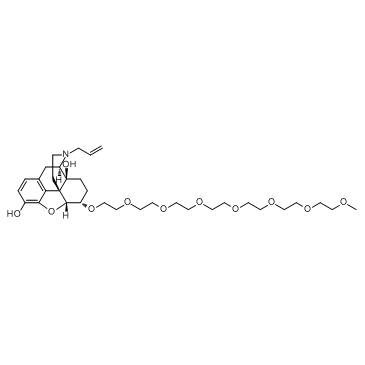
| DC20016 | Naloxegol Featured |
Naloxegol is a CYP3A4 enzyme inhibitor, is a peripherally-selective opioid antagonist, for the treatment of opioid-induced constipation.
More description
|
|
| DC9619 | Nafamostat (mesylate) Featured |
Nafamostat mesylate, a synthetic serine protease inhibitor, is an anticoagulant,showed highly potent activity against COVID-19(SARS-COV-2).
More description
|

|
| DC7471 | N6022 Featured |
N6022 is a potent, selective, reversible, and efficacious S-Nitrosoglutathione reductase(GSNOR) inhibitor(IC50=10 nM) which is currently undergoing clinical development.
More description
|

|
| DC9298 | thiotepa Featured |
N,N’N’-triethylenethiophosphoramide (ThioTEPA) is a cancer chemotherapeutic member of the alkylating agent group.
More description
|

|
| DC10074 | MX69 Featured |
MX69 is the MDM2/XIAP inhibitor, used for cancer treatment.
More description
|

|
| DC10871 | MTX211 Featured |
MTX-211 is a dual inhibitor of EGFR and PI3K, used for the treatment of cancer and other diseases.
More description
|

|
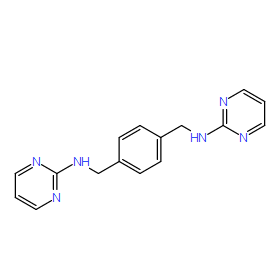
| DC8822 | MSX-122 Featured |
MSX-122 is a n orally bioavailable inhibitor of CXCR4 with potential antineoplastic and antiviral activities.
More description
|
|